Vyondys 53
Oplysningerne På Webstedet Er Ikke Medicinsk Rådgivning. Vi Sælger Ikke Noget. Nøjagtigheden Af Oversættelsen Er Ikke Garanteret. Ansvarsfraskrivelse
Resume af lægemiddeloversigt
Hvad er Vyondys 53?
Vyondys 53 (Golodirsen) er et antisense -oligonukleotid, der er indikeret til behandling af Duchenne muskeldystrofi (DMD) hos patienter, der har en bekræftet mutation af DMD -genet, der er tilgængelig for at ekson 53 springer over.
Hvad er bivirkninger af Vyondys 53?
Vyondys 53
- elveblest
- Besvær
- Hævelse af dine ansigtslæber tunge eller hals
- kløe
- udslæt
- Blærende eller skrælning af huden
- lyserød brun eller rød urin
- skummende urin og
- hævelse i dine ansigt hænder fødder eller mave
Få medicinsk hjælp med det samme, hvis du har nogen af de symptomer, der er anført ovenfor.
Bivirkninger af Vyondys 53 inkluderer:
- hovedpine
- feber
- falder
- mavesmerter
- løbende eller Snerpet næse
- hoste
- opkast
- kvalme
- Administrationsstedets smerte
- Rygsmerter
- diarre
- svimmelhed
- ligamentforstuvning
- Kontusion
- influenza
- Mund og hals smerte
- Hud slid
- Øreinfektion
- sæson- allergi
- Hurtig hjerterytme
- Kateterpladsrelateret reaktion
- forstoppelse og
- brud
Søg medicinsk behandling eller ring 911 på én gang, hvis du har følgende alvorlige bivirkninger:
- Alvorlige øjensymptomer såsom pludseligt synstab sløret synet tunnel vision øje smerter eller hævelse eller at se haloer omkring lys;
- Alvorlige hjertesymptomer såsom hurtig uregelmæssige eller bankende hjerteslag; flagrende i brystet; åndenød; Og pludselig svimmelhed Lightheadedness eller gå ud;
- Alvorlig hovedpine forvirring sløret talearm eller ben svaghed problemer med at gå tab af koordination føles ustabil meget stive muskler høj feber voldsom sved eller rysten.
Dette dokument indeholder ikke alle mulige bivirkninger, og andre kan forekomme. Kontakt din læge for yderligere oplysninger om bivirkninger.
Dosering til Vyondys 53
Dosis af Vyondys 53 er 30 milligram pr. Kg en gang ugentligt.
Vyondys 53 hos børn
Vyondys 53 er indikeret til behandling af Duchenne muskeldystrofi (DMD) hos patienter, der har en bekræftet mutation af DMD -genet, der er tilgængelig for at exon 53 springer inklusive pædiatriske patienter.
Hvilke stoffer stoffer eller kosttilskud interagerer med Vyondys 53?
Vyondys 53 kan interagere med andre lægemidler.
Fortæl din læge alle medicin og kosttilskud, du bruger.
Vyondys 53 under graviditet og amning
Fortæl din læge, hvis du er gravid eller planlægger at blive gravid, før du bruger Vyondys 53; Det er ukendt, hvordan det ville påvirke et foster. Det er ukendt, om Vyondys 53 passerer til modermælk. Kontakt din læge inden amning.
Yderligere oplysninger
Vores Vyondys 53 (Golodirsen) injektion til intravenøs brug bivirkninger Drug Center giver et omfattende overblik over tilgængelige lægemiddelinformation om de potentielle bivirkninger, når du tager denne medicin.
Dette er ikke en komplet liste over bivirkninger, og andre kan forekomme. Ring til din læge for medicinsk rådgivning om bivirkninger. Du kan rapportere bivirkninger til FDA på 1-800-FDA-1088.
FDA -lægemiddelinformation
- Lægemiddelbeskrivelse
- Indikationer
- Bivirkninger
- Advarsler
- Overdosis
- Klinisk farmakologi
- Medicin vejledning
Beskrivelse til Vyondys 53
Vyondys 53 (Golodirsen) injektion er en steril vandig konserveringsfri koncentreret opløsning til fortynding inden intravenøs administration. Vyondys 53 er en klar til lidt opalescent farveløs væske. Vyondys 53 leveres i enkeltdosis hætteglas indeholdende 100 mg golodirsen (50 mg/ml). Vyondys 53 er formuleret som en isotonisk phosphatbufret saltopløsning med en osmolalitet på 260 til 320 mosm og en pH på 7,5. Hver milliliter af Vyondys 53 indeholder: 50 mg golodirsen; 0,2 mg kaliumchlorid; 0,2 mg kaliumphosphat monobasisk; 8 mg natriumchlorid; og 1,14 mg natriumphosphatdibasisk vandfri i vand til injektion. Produktet kan indeholde saltsyre eller natriumhydroxid for at justere pH.
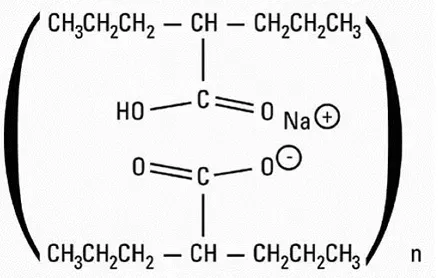
Golodirsen er et antisense -oligonukleotid af phosphorodiamidat -morpholino -oligomer (PMO) underklasse. PMO'er er syntetiske molekyler, hvor de fem-ledede ribofuranosylringe, der findes i naturligt DNA og RNA, erstattes af en seks-ledet morpholino-ring. Hver morpholino -ring er forbundet gennem en uladet phosphorodiamid -del snarere end den negativt ladede fosfatbinding, der er til stede i naturligt DNA og RNA. Hver phosphorodiamidat -morpholino -underenhed indeholder en af de heterocykliske baser, der findes i DNA (adenincytosin guanin eller thymin). Golodirsen indeholder 25 tilknyttede underenheder. Sekvensen af baser fra 5 'enden til 3' ende er gttgcctccggttctgaaggtgttc. Den molekylære formel af Golodirsen er C305H481N138O112P25 og molekylvægten er 8647,28 Daltons. Strukturen af Golodirsen er:
 |
Bruger til Vyondys 53
Vyondys 53 er indikeret til behandling af Duchenne muskeldystrofi (DMD) hos patienter, der har en bekræftet mutation af DMD -genet, der er tilgængelig for at ekson 53 springer over. This indication is approved under accelerated approval based on an increase in dystrophin production in skeletal muscle observed in patients treated with Vyondys 53 [see Kliniske studier ]. Fortsat godkendelse af denne indikation kan muligvis være betinget af verifikation af en klinisk fordel i bekræftende forsøg.
Dosering til Vyondys 53
Overvågning for at vurdere sikkerheden
Serumcystatin C urin-målepind og urinprotein-til-kreatininforhold skal måles, før Vyondys 53 startes. Få urinprøverne inden infusion af Vyondys 53 eller mindst 48 timer efter den seneste infusion [se Advarsler og forholdsregler ].
Doseringsoplysninger
Den anbefalede dosering af Vyondys 53 er 30 milligram pr. Kg indgivet en gang ugentligt som en 35 til 60-minutters intravenøs infusion via et in-line 0,2 mikronfilter.
Hvis en dosis Vyondys 53 går glip af, kan den administreres så hurtigt som muligt efter den planlagte dosis.
Forberedelsesinstruktioner
Vyondys 53 leveres i enkeltdosis hætteglas som en konserveringsfri koncentreret løsning, der kræver fortynding før administration. Parenterale lægemiddelprodukter skal inspiceres visuelt for partikler og misfarvning inden administration hver gang løsning og containertilladelse. Brug aseptisk teknik.
- Beregn den samlede dosis af Vyondys 53, der skal administreres baseret på patientens vægt og den anbefalede dosis på 30 milligram pr. Kg. Bestem mængden af Vyondys 53 nødvendige og det korrekte antal hætteglas for at levere den fulde beregnede dosis.
- Lad hætteglassene varme til stuetemperatur. Bland indholdet af hvert hætteglas ved forsigtigt at invertere 2 eller 3 gange. Ryst ikke.
- Undersøg visuelt hvert hætteglas af Vyondys 53. Opløsningen er en klar til lidt opalescent farveløs væske og kan indeholde spormængder af små hvide til off-white amorfe partikler. Brug ikke, hvis opløsningen i hætteglassene er overskyet misfarvet eller indeholder andet partikler bortset fra spormængder af små hvide til off-white amorfe partikler.
- Med en sprøjte udstyret med en 21-gauge eller mindre boring af ikke-korende nål trækker det beregnede volumen af Vyondys 53 tilbage fra det passende antal hætteglas.
- Fortynd den tilbagetrukne Vyondys 53 i 0,9% natriumchloridinjektion USP for at fremstille et samlet volumen på 100 til150 ml. Inverter forsigtigt 2 til 3 gange for at blande. Ryst ikke. Undersøg visuelt den fortyndede løsning. Brug ikke, hvis opløsningen er overskyet misfarvet eller indeholder andre fremmede partikler end spormængder af små hvide til off-white amorfe partikler.
- Administrer den fortyndede opløsning via et in-line 0,2 mikronfilter.
- Vyondys 53 indeholder ingen konserveringsmidler og bør administreres umiddelbart efter fortynding. Komplet infusion af fortyndet Vyondys 53 inden for 4 timer efter fortynding. Hvis øjeblikkelig anvendelse ikke er muligt, kan det fortyndede produkt opbevares i op til 24 timer ved 2 ° C til 8 ° C (36 ° F til 46 ° F). Frys ikke. Kasser ubrugt Vyondys 53.
Administrationsinstruktioner
Anvendelse af en aktuel bedøvelsescreme på infusionsstedet inden administration af Vyondys 53 kan overvejes.
Vyondys 53 administreres via intravenøs infusion. Skyl den intravenøse adgangslinie med 0,9% natriumchloridinjektion USP før og efter infusion.
Tilfør den fortyndede Vyondys 53 over 35 til 60 minutter via et in-line 0,2 mikronfilter. Bland ikke andre medicin med Vyondys 53 eller tilfør andre medicin samtidig via den samme intravenøse adgangslinje med Vyondys 53.
Hvis der opstår en overfølsomhedsreaktion, skal du overveje at bremse infusionen, der afbryder eller afbryder Vyondys 53 -terapi [se Kontraindikationer Advarsler og forholdsregler og Bivirkninger ].
Hvor leveret
Doseringsformularer og styrker
Vyondys 53 er en klar til lidt opalescent farveløs væske og kan indeholde spormængder af små hvide til off-white amorfe partikler og fås som:
- Indsprøjtning : 100 mg/2 ml (50 mg/ml) opløsning i en enkeltdosis hætteglas
Vyondys 53 Injektion leveres i enkeltdosis hætteglas. Løsningen er en klar til lidt opalescent farveløs væske og kan indeholde spormængder af små hvide til off-hvide amorfe partikler.
Enkeltdosis hætteglas, der indeholder 100 mg/2 ml (50 mg/ml) NDC 60923-465-02
Opbevaring og håndtering
Opbevar Vyondys 53 ved 2 ° C til 8 ° C (36 ° F til 46 ° F). Frys ikke. Opbevares i original karton, indtil den er klar til brug til beskyttelse mod lys.
Fremstillet til: Sarepta Therapeutics Inc. Cambridge MA 02142 USA. Revideret: juni 2024
Bivirkninger til Vyondys 53
Følgende alvorlige bivirkninger er beskrevet nedenfor og andre steder i mærkningen:
- Overfølsomhedsreaktioner [se Advarsler og forholdsregler ]
Kliniske forsøg oplever
Fordi kliniske forsøg udføres under vidt forskellige tilstande, kan der ikke sammenlignes bivirkninger, der er observeret i de kliniske forsøg med et lægemiddel, ikke direkte med hastigheder i de kliniske forsøg med et andet lægemiddel og muligvis ikke afspejler de satser, der er observeret i praksis.
I Vyondys 53 klinisk udviklingsprogram modtog 58 patienter mindst en intravenøs dosis af Vyondys 53 mellem 4 mg/kg (0,13 gange den anbefalede dosering) og 30 mg/kg (den anbefalede dosering). Alle patienter var mandlige og havde genetisk bekræftet Duchenne muskeldystrofi. Alder ved studieindgangen var 6 til 13 år. De fleste (86%) patienter var kaukasiske.
Vyondys 53 was studied in 2 double-blind placebo-controlled studies.
I undersøgelse 1 del 1 blev patienter randomiseret til at modtage en gang om ulige intravenøse infusioner af Vyondys 53 (n = 8) i fire stigende dosisniveauer fra 4 mg/kg til 30 mg/kg eller placebo (n = 4) i mindst 2 uger på hvert niveau. Alle patienter, der deltog i undersøgelse 1 del 1 (n = 12) Kliniske studier ].
Hos undersøgelse modtog 2 patienter Vyondys 53 (n = 33) 30 mg/kg eller placebo (n = 17) IV en gang ugentligt i op til 96 uger, hvorefter alle patienter modtog Vyondys 53 i en dosis på 30 mg/kg.
Bivirkninger observeret hos mindst 20% af de behandlede patienter i de placebokontrollerede sektioner af undersøgelser 1 og 2 er vist i tabel 1.
Tabel 1: Bivirkninger, der forekom hos mindst 20% af Vyondys 53-behandlede patienter og med en hastighed, der var større end placebo i undersøgelser 1 og 2
| Bivirkning | Vyondys 53 (N = 41) % | Placebo (N = 21) % |
| Hovedpine | 41 | 10 |
| Pyrexia | 41 | 14 |
| Falde | 29 | 19 |
| Mavesmerter | 27 | 10 |
| Nasopharyngitis | 27 | 14 |
| Hoste | 27 | 19 |
| Opkast | 27 | 19 |
| Kvalme | 20 | 10 |
Andre bivirkninger, der opstod ved en frekvens større end 5% af Vyondys 53-behandlede patienter og ved en større frekvens end placebo var: Administrationsstedets smerte rygsmerter Smerter diarré Dizziness Ligament Forstuvning Kontusion Influenza Oropharyngeal Pain Rhinitis Skin Abrasion Ear infektion sæsonbestemt allergi Tachycardia kateter -relateret reaktionsforstoppelse og brud.
Overfølsomhedsreaktioner har fundet sted hos patienter behandlet med Vyondys 53 [se Advarsler og forholdsregler ].
svovl hvad bruges det til
Oplevelse af postmarketing
Følgende bivirkninger er blevet identificeret under postapproval brug af Vyondys 53. Fordi disse reaktioner rapporteres frivilligt fra en population af usikker størrelse, er det ikke altid muligt at pålideligt estimere deres frekvens eller etablere et årsagsforhold til lægemiddeleksponering.
Immunsystemforstyrrelser: Anafylaksi [se Kontraindikationer og Advarsler og forholdsregler ]
Lægemiddelinteraktioner for Vyondys 53
Ingen oplysninger leveret
Advarsler for Vyondys 53
Inkluderet som en del af FORHOLDSREGLER afsnit.
Forholdsregler for Vyondys 53
Overfølsomhedsreaktioner
Overfølsomhedsreaktioner, herunder anafylaksi-udslæt pyrexia pruritus urticaria dermatitis og hud-eksfoliering, har forekommet hos Vyondys 53-behandlede patienter, der kræver behandling. Hvis der opstår en overfølsomhedsreaktion, der opstår, skal du institut Dosering og administration ]. Vyondys 53 is contraindicated in patients with a history of a serious hypersensitivity reaction to golodirsen or to any of the inactive ingredients in Vyondys 53 [see Kontraindikationer ].
Nyretoksicitet
Nyretoksicitet blev observeret hos dyr, der modtog Golodirsen [se Brug i specifikke populationer ]. Although kidney toxicity was not observed in the clinical studies with Vyondys 53 the clinical experience with Vyondys 53 is limited og kidney toxicity including potentially fatal glomerulonephritis has been observed after administration of some antisense oligonucleotides. Kidney function should be monitored in patients taking Vyondys 53. Because of the effect of reduced skeletal muscle mass on creatinine measurements creatinine may not be a reliable measure of kidney function in DMD patients. Serum cystatin C urine dipstick og urine protein-to-creatinine ratio should be measured before starting Vyondys 53. Consider also measuring glomerular filtration rate using an exogenous filtration marker before starting Vyondys 53. During treatment monitor urine dipstick every month og serum cystatin C og urine protein-to-creatinine ratio every three months. Only urine expected to be free of excreted Vyondys 53 should be used for monitoring of urine protein. Urine obtained on the day of Vyondys 53 infusion prior to the infusion or urine obtained at least 48 hours after the most recent infusion may be used. Alternatively use a laboratory test that does not use the reagent pyrogallol red as this reagent has the potential to cross react with any Vyondys 53 that is excreted in the urine og thus lead to a false positive result for urine protein.
Hvis en vedvarende stigning i serumcystatin C eller proteinuri påvises, henvises til en pædiatrisk nefrolog for yderligere evaluering.
Ikke -klinisk toksikologi
Karcinogenese mutagenese nedskrivning af fertilitet
Carcinogenese
Administration af Golodirsen til mandlige transgene (Tg.Rash2) mus (0 100 300 eller 1000 mg/kg) ugentligt i 26 uger ved subkutan injektion og til hanrotter (0 30 100 eller 300 mg/kg) ugentligt i op til 102 uger ved intravenøs injektion resulterede i ingen stigning i neoplasmer.
Mutagenese
Golodirsen var negativ i in vitro (bakteriel omvendt mutation og kromosomal afvigelse i CHO -celler) og in vivo (mus knoglemarv mikronukleus) assays.
Værdiforringelse af fertiliteten
Fertilitetsundersøgelser hos dyr blev ikke udført med Golodirsen. Ingen effekter af Golodirsen på det mandlige reproduktive system blev observeret efter ugentlig subkutan administration (0 120 300 eller 600 mg/kg til mandlige mus eller ugentlige intravenøse administration (0 80 200 eller 400 mg/kg) til mandlige aber. Plasma -eksponering (AUC) ved de højeste doser, Dosis på 30 mg/kg.
Brug i specifikke populationer
Graviditet
Risikooversigt
Der er ingen mennesker- eller dyredata til rådighed for at vurdere brugen af Vyondys 53 under graviditet. I den amerikanske generelle befolkning forekommer der store fødselsdefekter i 2 til 4%, og spontanabort forekommer i 15 til 20% af klinisk anerkendte graviditeter.
Amning
Risikooversigt
Der er ingen menneskelige eller dyredata til at vurdere virkningen af Vyondys 53 på mælkeproduktionen tilstedeværelsen af Golodirsen i mælk eller virkningerne af Vyondys 53 på det ammede spædbarn.
De udviklingsmæssige og sundhedsmæssige fordele ved amning bør overvejes sammen med mors kliniske behov for Vyondys 53 og eventuelle bivirkninger på det ammede spædbarn fra Vyondys 53 eller fra den underliggende moderlige tilstand.
Pædiatrisk brug
Vyondys 53 er indikeret til behandling af Duchenne muskeldystrofi (DMD) in patients who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping including pediatric patients [see Kliniske studier ].
Intravenøs administration af Golodirsen (0 100 300 eller 900 mg/kg) til unge mandlige rotter en gang ugentligt i 10 uger (postnatal dage 14 til 77) resulterede ikke i postnatal udviklingsmæssig (f.eks. Neurobehavioral immunfunktion eller mandlig reproduktiv) toksicitet. Imidlertid resulterede Golodirsen ved den højeste dosis (900 mg/kg/uge) golodirsen i dyrs død på grund af nedsat nyrefunktion eller fiasko. Hos overlevende dyr (inklusive et dyr i den laveste testede dosis) var der en dosisafhængig stigning i forekomsten og sværhedsgraden af nyrerørformede effekter (inklusive degeneration/regenereringsfibrose-vakuolering og dilatation), som korrelerede med ændringer i kliniske patologiparametre, der reflekterede en dosisafhængig svækkelse af nyrefunktionen. Derudover blev der observeret fald i knoglerområdet mineralindhold og mineraltæthed i den højeste testede dosis (900 mg/kg uge), men uden effekt på knoglevækst. En dosis uden virkning til nyretoksicitet blev ikke identificeret; Den laveste testede dosis (100 mg/kg/uge) var forbundet med plasmaeksponeringer (AUC) ca. 2,5 gange, at hos mennesker ved den anbefalede humane dosis på 30 mg/kg/uge.
Geriatrisk brug
DMD er stort set en sygdom hos børn og unge voksne; Derfor er der ingen geriatrisk oplevelse med Vyondys 53.
Patienter med nedsat nyrefunktion
Nyreafstand af Golodirsen reduceres hos ikke-DMD-voksne med nedsat nyrefunktion baseret på estimeret glomerulær filtreringshastighed beregnet ved hjælp af modifikation af diæt og nyresygdom (MDRD) ligning [Se Klinisk farmakologi ]. However because of the effect of reduced skeletal muscle mass on creatinine measurements in DMD patients no specific dosage adjustment can be recommended for DMD patients with renal impairment based on estimated glomerular filtration rate. Patients with known renal function impairment should be closely monitored during treatment with Vyondys 53.
Overdoseringsoplysninger til Vyondys 53
Ingen oplysninger leveret
Kontraindikationer for Vyondys 53
Vyondys 53 is contraindicated in patients with a serious hypersensitivity reaction to golodirsen or to any of the inactive ingredients in Vyondys 53. Anaphylaxis has occurred in patients receiving Vyondys 53 [see Advarsler og forholdsregler ].
Klinisk farmakologi for Vyondys 53
Handlingsmekanisme
Golodirsen er designet til at binde til exon 53 af dystrophin pre-mRNA, hvilket resulterer i udelukkelse af denne exon under mRNA-behandling hos patienter med genetiske mutationer, der er tilgængelige for at exon 53 springer over. Exon 53 Spring er beregnet til at muliggøre produktion af et internt trunkeret dystrophinprotein hos patienter med genetiske mutationer, der er tilgængelige for at ekson 53 springer over [Se Kliniske studier ].
Farmakodynamik
Efter behandling med Vyondys 53 vurderede alle patienter (n = 25) i undersøgelse 1 del 2 [se Kliniske studier ] havde en stigning i spring over exon 53 demonstreret ved omvendt transkriptionspolymerasekædereaktion (RT-PCR) sammenlignet med baseline.
I undersøgelse 1 del 2 [se Kliniske studier ] Dystrophinniveauer som vurderet af Sarepta Western blot -assayet steg fra 0,10% (SD 0,07) normal ved baseline til 1,02% (SD 1,03) af normal efter 48 ugers behandling med Vyondys 53. Den gennemsnitlige ændring fra baseline i dystrophin efter 48 ugers behandling med Vyondys 53 var 0,92% (SD 1,01) af normale niveauer (Pystrophin<0.001); the median change from baseline was 0.88%. This increase in dystrophin protein expression positively correlated with the level of exon skipping. Dystrophin levels assessed by western blot can be meaningfully influenced by differences in sample processing analytical technique reference materials og quantitation methodologies. Therefore comparing dystrophin results from different assay protocols will require a stogardized reference material og additional bridging studies.
Korrekt lokalisering af trunkeret dystrophin til sarkolemmaet i muskelfibre hos patienter behandlet med Golodirsen blev demonstreret ved immunofluorescensfarvning.
Farmakokinetik
Farmakokinetikken af Golodirsen blev evalueret hos DMD -patienter efter administration af intravenøse doser, der spænder fra 4 mg/kg/uge til 30 mg/kg/uge (dvs. anbefalet dosering). Golodirsen-eksponering steg proportionalt med dosis med minimal akkumulering med en gang om ugen dosering. Inter-subjektvariabilitet (som% CV) for Cmax og AUC varierede fra henholdsvis 38% til 72% og 34% til 44%.
Fordeling
Stadet tilstand af distributionsmængde var ens mellem DMD-patienter og raske forsøgspersoner. Den gennemsnitlige Golodirsen-steady-state-mængde af distribution var 668 ml/kg (%CV = 32,3) i en dosis på 30 mg/kg. Golodirsen Plasma -proteinbinding varierede fra 33% til 39% og er ikke koncentrationsafhængig.
Eliminering
Golodirsen eliminering af halveringstid (SD) var 3,4 (0,6) timer, og plasmaklarering var 346 ml/t/kg ved dosis på 30 mg/kg.
Metabolisme
Golodirsen er metabolisk stabil. Ingen metabolitter blev påvist i plasma eller urin.
Udskillelse
Golodirsen udskilles for det meste uændret i urinen. Eliminationshalveringstiden (T½) var 3,4 timer.
Specifikke populationer
Alder
Farmakokinetikken af Golodirsen er blevet evalueret hos mandlige pædiatriske DMD -patienter. Der er ingen erfaring med brugen af Vyondys 53 hos DMD -patienter 65 år eller ældre.
Køn
Køn effects have not been evaluated; Vyondys 53 has not been studied in female patients.
Race
Den potentielle virkning af race er ikke kendt, fordi 92% af patienterne i undersøgelser var kaukasiere.
Patienter med nedsat nyrefunktion
Effekten af nedsat nyrefunktion på farmakokinetikken af Golodirsen blev evalueret hos ikke-DMD-personer i alderen 41 til 65 år med trin 2 kronisk nyresygdom (CKD) (N = 8 estimeret glomerulær filtreringshastighed (EGFR) ≥60 og <90 mL/min/1.73 m²) or Stage 3 CKD (n=8 eGFR ≥30 og <60 mL/min/1.73 m²) og matched healthy subjects (n=8 eGFR ≥90 mL/min/1.73 m²). Subjects received a single 30 mg/kg IV dose of golodirsen.
Hos forsøgspersoner med trin 2 eller fase 3 CKD-eksponering (AUC) steg henholdsvis ca. 1,2 gange og 1,9 gange. Der var ingen ændring i Cmax hos personer med trin 2 CKD; Hos personer med trin 3 CKD var der en 1,2 gange stigning i Cmax sammenlignet med personer med normal nyrefunktion. Effekten af trin 4 eller trin 5 CKD på Golodirsen farmakokinetik og sikkerhed er ikke undersøgt.
Estimerede GFR -værdier afledt af MDRD -ligninger og tærskeldefinitionerne for forskellige CKD -stadier hos ellers ville raske voksne ikke kunne generaliseres for pædiatriske patienter med DMD. Derfor kan ingen specifik doseringsjustering anbefales til patienter med nedsat nyrefunktion [se Brug i specifikke populationer ].
Patienter med nedsat leverfunktion:
Vyondys 53 has not been studied in patients with hepatic impairment.
Lægemiddelinteraktionsundersøgelser
Golodirsen hæmmede ikke CYP1A2 CYP2B6 CYP2C8 CYP2C9 CYP2C19 CYP2D6 eller CYP3A4/5 in vitro. Golodirsen var en svag inducer af CYP1A2 og inducerede ikke CYP2B6 eller CYP3A4. Golodirsen blev ikke metaboliseret af humane levermikrosomer og var ikke et substrat eller en stærk inhibitor af nogen af de vigtigste testede humane lægemiddeltransportører (OAT1 OAT3 OCT2 OATP1B1 MATE1 P-GP BCRP og MRP2 OATP1B3 og MATE2-K). Baseret på in vitro-data har Golodirsen et lavt potentiale for medikament-lægemiddelinteraktioner hos mennesker.
Dyretoksikologi og/eller farmakologi
Nyretoksicitet blev observeret i undersøgelser i mandlige mus og rotter; Resultater i urinblære blev observeret hos hanmus.
Hos mandlige mus blev Golodirsen administreret ugentligt i 12 uger ved intravenøs injektion (0 12 120 eller 960 mg/kg) eller i 26 uger ved subkutan injektion (0 120 300 eller 600 mg/kg). I de 12-ugers undersøgelsesmikroskopiske fund i nyre (rørformet dilatationsbasofil eller eosinofil kastes vakuolering) korrelerede med stigninger i serummarkører af nyrefunktion (f.eks. Urea Nitrogenkreatinin) blev primært observeret ved den højeste testede dosis; Hypertrofi af det overgangsepitel i ureter eller urinblære blev observeret i alle doser. I den 26-ugers undersøgelse blev der observeret renal tubulær degeneration og degeneration af overgangsepitelet i urinblæren i alle doser.
har Spiriva steroider i det
Hos mandlige rotter resulterede intravenøs administration af Golodirsen (0 60 100 300 eller 600 mg/kg) ugentligt i 13 uger i rørformet degeneration overhovedet, men den laveste testede dosis; Ved den høje dosis blev de mikroskopiske ændringer ledsaget af stigninger i serumurinstofnitrogen.
I mandlige aber, der blev intravenøs administration af Golodirsen (0 80 200 eller 400 mg/kg) ugentligt i 39 uger, resulterede i mikroskopiske ændringer i nyre (basofilia -dilatation eller mononuklear celleinfiltration) ved alle doser, der korrelerede med stigninger i serummarkører af nøjefunktion (uren nitrogen -skabelse af højest dosen.
Kliniske studier
Effekten af Vyondys 53 på dystrophinproduktion blev evalueret i en undersøgelse hos DMD -patienter med en bekræftet mutation af DMD -genet, der er tilgængelig for at exon 53 spring (undersøgelse 1; NCT02310906).
Undersøgelse 1 del 1 var en dobbeltblind placebokontrolleret dosis-titreringsundersøgelse hos 12 DMD-patienter. Patienter blev randomiseret 2: 1 for at modtage Vyondys 53 eller matchende placebo. Vyondys 53âbehandlede patienter modtog fire eskalerende dosisniveauer, der spænder fra 4 mg/kg/uge (mindre end den anbefalede dosering) til 30 mg/kg/uge ved intravenøs infusion i 2 uger på hvert dosisniveau.
Undersøgelse 1 del 2 var en 168-ugers open-label-undersøgelse, der vurderede effektiviteten og sikkerheden af Vyondys 53 i en dosis på 30 mg/kg/uge i de 12 patienter, der var indskrevet i del 1 plus 13 yderligere behandlingsnaive patienter med DMD AMENABLE til exon 53, der springer over. Ved undersøgelsesindgangen (enten i del 1 eller del 2) havde patienter en medianalder på 8 år og var på en stabil dosis af kortikosteroider i mindst 6 måneder. Efficacy was assessed based on change from baseline in the dystrophin protein level (measured as % of the dystrophin level in healthy subjects i.e. % of normal) at Week 48 of Part 2. Muscle biopsies were obtained at baseline prior to treatment and at Week 48 of Part 2 in all VYONDYS 53-treated patients (n=25) and were analyzed for dystrophin protein level by Sarepta western blot. Gennemsnitlige dystrophinniveauer steg fra 0,10% (SD 0,07) normal ved baseline til 1,02% (SD 1,03) af normal i uge 48 i undersøgelse 1 del 2 med en gennemsnitlig ændring i dystrophin på 0,92% (SD 1,01) af normale niveauer (P<0.001); the median change from baseline was 0.88%.
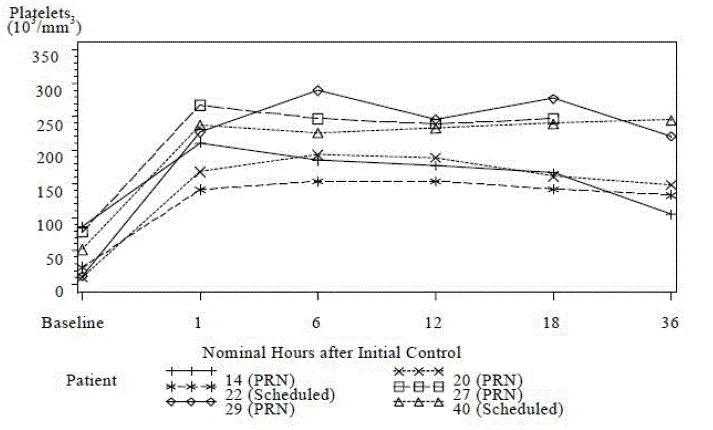
Individuelle patientdystrophinniveauer fra undersøgelse 1 er vist i tabel 2.
Tabel 2: Dystrophinekspression Sarepta Western blot af individuel patient fra undersøgelse 1
| Patientnummer | Sarepta western blot % normal dystrophin | Patientnummer | Sarepta western blot % normal dystrophin | ||||
| Baseline | Del 2 uge 48 | Skift fra baseline | Baseline | Del 2 uge 48 | Skift fra baseline | ||
| 1 | 0.08 | 0.09 | 0.01 | 14 | 0.22 | 0.28 | 0.06 |
| 2 | 0.11 | 0.11 | 0.01 | 15 | 0.14 | 0.21 | 0.07 |
| 3 | 0.21 | 0.22 | 0.01 | 16 | 0.05 | 0.42 | 0.37 |
| 4 | 0.05 | 0.12 | 0.08 | 17 | 0.07 | 1.03 | 0.97 |
| 5 | 0.03 | 0.12 | 0.09 | 18 | 0.02 | 1.57 | 1.55 |
| 6 | 0.06 | 0.14 | 0.09 | 19 | 0.12 | 1.17 | 1.05 |
| 7 | 0.12 | 0.37 | 0.25 | 20 | 0.03 | 1.72 | 1.69 |
| 8 | 0.11 | 1.06 | 0.95 | 21 | 0.11 | 1.77 | 1.66 |
| 9 | 0.06 | 0.54 | 0.48 | 22 | 0.31 | 4.30 | 3.99 |
| 10 | 0.05 | 0.97 | 0.92 | 23 | 0.11 | 0.36 | 0.25 |
| 11 | 0.06 | 1.55 | 1.49 | 24 | 0.03 | 0.91 | 0.88 |
| 12 | 0.07 | 1.91 | 1.84 | 25 | 0.07 | 1.29 | 1.22 |
| 13 | 0.10 | 3.25 | 3.15 |
Patientinformation til Vyondys 53
Overfølsomhedsreaktioner
Rådgiv patienter og/eller plejepersonale om, at overfølsomhedsreaktioner, herunder anafylakse udslæt pyrexia kløe urticaria dermatitis og hudeksfoliering, har fundet sted hos patienter, der blev behandlet med Vyondys 53. Instruer dem til at søge øjeblikkelig medicinsk behandling, hvis de oplever tegn og symptomer på hypersitivitet [se Advarsler og forholdsregler ].
Nyretoksicitet
Informer patienter Nefrotoksicitet er forekommet med lægemidler, der ligner Vyondys 53. Rådgiv patienter om vigtigheden af overvågning for nyretoksicitet fra deres sundhedsudbydere under behandling med Vyondys 53 [se Advarsler og forholdsregler ].