Ultomiris
Oplysningerne På Webstedet Er Ikke Medicinsk Rådgivning. Vi Sælger Ikke Noget. Nøjagtigheden Af Oversættelsen Er Ikke Garanteret. Ansvarsfraskrivelse
Resume af lægemiddeloversigt
Hvad er Ultomiris?
Ultomiris (Ravulizumab-CWVZ) er en komplementinhibitor, der er angivet til behandling af voksne patienter med paroxysmal natlig Hæmoglobinuri (PNH).
Hvad er bivirkninger af ultomiris?
Ultomiris
Hvad gør beta alaninpulver
- elveblest
- Besvær
- Hævelse af dine ansigtslæber tunge eller hals
- Lightheadedness
- brystsmerter
- Muskelsmerter med influenzalignende symptomer
- feber med udslæt
- Feber med hovedpine
- hovedpine og stivhed i din hals eller ryg
- hovedpine med kvalme eller opkast
- forvirring
- Dine øjne er måske mere følsomme over for lys
- Smerter eller brændende, når du tisser
- Smerter eller hævelse af kønsorganet eller rektal areal
- Usædvanlig vaginal blødning
- Foul decharge fra penis eller vagina
- træthed
- forvirring
- mavesmerter
- problemer med at sluge
- Problemer med at have en erektion
- blod i din urin
- anfald og
- Tab af bevidsthed
Få medicinsk hjælp med det samme, hvis du har nogen af de symptomer, der er anført ovenfor.
Almindelige bivirkninger af ultomiris inkluderer:
- Øvre luftvejsinfektion
- hovedpine
- diarre
- kvalme
- mavesmerter
- feber
- Smerter i ekstremiteter
- ledssmerter og
- svimmelhed
Søg medicinsk behandling eller ring 911 på én gang, hvis du har følgende alvorlige bivirkninger:
- Alvorlige øjensymptomer såsom pludseligt synstab sløret synet tunnel vision øje smerter eller hævelse eller at se haloer omkring lys;
- Alvorlige hjertesymptomer såsom hurtig uregelmæssige eller bankende hjerteslag; flagrende i brystet; åndenød; og pludselig svimmelhed lethed eller udlevering;
- Alvorlig hovedpine forvirring sløret talearm eller ben svaghed problemer med at gå tab af koordination føles ustabil meget stive muskler høj feber voldsom sved eller rysten.
Dette dokument indeholder ikke alle mulige bivirkninger, og andre kan forekomme. Kontakt din læge for yderligere oplysninger om bivirkninger.
Dosering til ultomiris
Den anbefalede doseringsregime af Ultomiris til voksne patienter med PNH består af en belastningsdosis efterfulgt af vedligeholdelsesdosering, der er administreret ved intravenøs infusion. Doser administreres baseret på patientens kropsvægt.
Hvilke stoffer stoffer eller kosttilskud interagerer med ultomiris?
Ultomiris kan interagere med andre lægemidler. Fortæl din læge alle medicin og kosttilskud, du bruger.
Ultomiris under graviditet og amning
Fortæl din læge, hvis du er gravid eller planlægger at blive gravid, før du bruger Ultomiris; Det er ukendt, hvordan det ville påvirke et foster. Der er risici for moderen og fosteret forbundet med ubehandlet paroxysmal natlig hæmoglobinuri (PNH) under graviditet. Det er ukendt, om ultomiris passerer ind i modermælk. På grund af potentialet for alvorlige bivirkninger i et sygeplejebarn, der ammes, bør der afbrydes under behandling med Ultomiris og i 8 måneder efter den endelige dosis.
Yderligere oplysninger
Vores Ultomiris (Ravulizumab-CWVZ) injektion til intravenøs brug bivirkninger Drug Center giver et omfattende overblik over tilgængelige lægemiddelinformation om de potentielle bivirkninger, når du tager denne medicin.
FDA -lægemiddelinformation
- Lægemiddelbeskrivelse
- Indikationer
- Dosering
- Bivirkninger
- Advarsler
- Overdosis
- Klinisk farmakologi
- Medicin vejledning
ADVARSEL
Alvorlige meningokokkinfektioner
- Livstruende meningokokkinfektioner/sepsis har forekommet hos patienter behandlet med ultomiris. Meningococcal-infektion kan blive hurtigt livstruende eller dødelig, hvis ikke anerkendt og behandlet tidligt [se advarsler og FORHOLDSREGLER ].
- Overhold det mest nuværende rådgivende udvalg for immuniseringspraksis (ACIP) henstillinger til meningokokkvaccination hos patienter med komplementmangel.
- Immuniser patienter med meningokokkvacciner mindst 2 uger før administration af den første dosis af ultomiris, medmindre risikoen for at forsinke Ultomiris -terapi opvejer risikoen for at udvikle en meningokokkinfektion [se advarsler og advarsler og advarsler og advarsler og advarsler og advarsler og advarsler og advarsler FORHOLDSREGLER for yderligere vejledning om håndtering af risikoen for meningokokkinfektion].
- Vaccination reduceres, men eliminerer ikke risikoen for meningokokkinfektioner. Overvåg patienter for tidlige tegn på meningokokkinfektioner og evaluer øjeblikkeligt, om der er mistanke om infektion.
Ultomiris er kun tilgængelig gennem et begrænset program under en risikovurderings- og afbødningsstrategi (REMS). Under Ultomiris REMS -receptpligtige skal tilmelde sig programmet [se advarsler og FORHOLDSREGLER ]. Enrollment in the Ultomiris REMS program og additional infellermation are available by telephone: 1-844-259-6783 eller at www.ultomirisrems.com.
Beskrivelse for ultomiris
Ravulizumab-CWVZ En komplementinhibitor er et humaniseret monoklonalt antistof (MAB) produceret i kinesiske hamster-ovarieceller (CHO) celler. Ravulizumab-CWVZ består af 2 identiske 448 aminosyrens tunge kæder og 2 identiske 214 aminosyrelysekæder og har en molekylvægt på ca. 148 kDa. De konstante regioner af Ravulizumabcwvz inkluderer den humane Kappa Light Chain Constant Region og proteinet konstrueret IgG2/4 tung kæde konstant region.
Den tunge kæde CH1 -domæne -hængselsregion og de første 5 aminosyrer i CH2 -domænet matcher det humane IgG2 -aminosyresekvensrester 6 til 36 i CH2 -regionen (fælles for både human IgG2 og IgG4 aminosyresekvenser), mens resten af CH2 -domænet og CH3 -domænet matcher den humane IgG4 -aminosyresekvens. De tunge og lette kæde-variable regioner, der danner det humane C5-bindingssted, består af humane rammeregioner podet til murine komplementaritetsbestemmende regioner.
Ultomiris (Ravulizumab-CWVZ) injektion er en steril klar for gennemskinnelig let hvidlig farvekonserveringsfri opløsning til intravenøs brug. Hvert enkeltdosis hætteglas indeholder 300 mg ravulizumab-CWVZ i en koncentration på 10 mg/ml med en pH på 7,0. Hver ML indeholder også polysorbat 80 (0,2 mg) (vegetabilsk oprindelse) natriumchlorid (8,77 mg) natriumphosphatdibasisk (1,78 mg) natriumphosphatmonobasisk (0,46 mg) og vand til injektion USP.
Anvendelser til ultomiris
Paroxysmal natlig hæmoglobinuri
Ultomiris er indikeret til behandling af voksne og pædiatriske patienter en måned og ældre med paroxysmal natlig hæmoglobinuri (PNH).
Atypisk hæmolytisk uremisk syndrom
Ultomiris er indikeret til behandling af voksne og pædiatriske patienter en måned og ældre med atypisk hæmolytisk uremisk syndrom (AHUS) for at hæmme komplementmedieret thrombotisk mikroangiopati (TMA).
Begrænsninger af brug
Ultomiris er ikke indikeret til behandling af patienter med shiga-toksin E. coli-relateret hæmolytisk uremisk syndrom (STEC-Hus).
Generaliserede Myasthenia Gravis
Ultomiris er indikeret til behandling af voksne patienter med generaliseret myasthenia gravis (GMG), som er anti-acetylcholinreceptor (AChR) antistof-positive.
Neuromyelitis Optica Spectrum Disorder
Ultomiris er indikeret til behandling af voksne patienter med neuromyelitis optica -spektrumforstyrrelse (NMOSD), som er anti -aquaporin -4 (AQP4) antistofpositiv.
Dosering til ultomiris
Vigtige doseringsoplysninger
Ultomiris er kun beregnet til at blive administreret som en intravenøs infusion hos voksne eller pædiatriske patienter en måned og ældre.
Anbefalet vaccination og profylakse til meningokokkinfektion
Vacciner patienter mod meningokokkinfektion (serogrupper a c w y og b) i henhold til de nuværende ACIP -anbefalinger mindst 2 uger før påbegyndelse af ultomiris [se ADVARSELS AND FORHOLDSREGLER ].
Hvis presserende ultomiris -terapi er indikeret hos en patient, der ikke er ajour med meningokokkvacciner i henhold til ACIP -anbefalinger, giver patienten antibakteriel medikamentprofylakse og administrerer disse vacciner så hurtigt som muligt.
Udbydere af sundhedsydelser, der ordinerer ultomiris, skal tilmelde sig Ultomiris og Soliris Rems [se ADVARSELS AND FORHOLDSREGLER ].
Anbefalet dosering til intravenøs administration hos voksne og pædiatriske patienter med PNH eller AHUS og hos voksne patienter med GMG eller NMOSD
Den anbefalede intravenøse (IV) Ultomiris -belastning og vedligeholdelsesdosering hos voksne og pædiatriske patienter en måned eller ældre, der vejer 5 kg eller mere med PNH eller AHUS eller hos voksne patienter med GMG eller NMOSD, der vejer 40 kg eller større, er baseret på patientens kropsvægt som vist i tabel 1 med vedligeholdelsesdoser administreret hver 4 eller 8 uger fra 2 uger efter belastning af dosen.
IV -doseringsplanen må lejlighedsvis variere inden for 7 dage efter den planlagte infusionsdag (undtagen den første vedligeholdelsesdosis af Ultomiris); Men efterfølgende doser skal administreres i henhold til den oprindelige tidsplan.
Efter en ubesvaret IV Ultomiris dosis skulle patienten straks kontakte deres sundhedsudbyder.
Tabel 1: IV Administration af Ultomiris Vægtbaseret doseringsregime-PNH AHUS GMG eller NMOSD*
| Indikationer | Kropsvægtområde (kg) | Indlæsning af dosis (mg) ** | Vedligeholdelsesdosis (MG) og doseringsinterval | |
| Pnh eller ahus | 5 til mindre end 10 | 600 | 300 | Hver 4. uge |
| 10 til mindre end 20 | 600 | 600 | ||
| 20 til mindre end 30 | 900 | 2100 | Hver 8. uge | |
| 30 til mindre end 40 | 1200 | 2700 | ||
| Pnh ahus gmg eller nmosd | 40 til mindre end 60 | 2400 | 3000 | Hver 8. uge |
| 60 til mindre end 100 | 2700 | 3300 | ||
| 100 eller mere | 3000 | 3600 | ||
| *Se tabel 4 Tabel 5 Tabel 7 og tabel 8 for valg af den rette samlede volumen og maksimal infusionshastighed [Se Dosering og administration ] ** Se tabel 2 for Ultomiris -behandlingsinitieringsinstruktion og timing af belastningsdosis og vedligeholdelsesdosis |
Se tabel 2 for behandlingsinitieringsinstruktioner hos patienter, der er komplementinhibitorens behandlingsnaive eller skiftende behandling fra eculizumab.
Tabel 2: IV Administration af Ultomiris -behandlingsinitieringsinstruktioner - PNH AHUS GMG eller NMOSD
| Befolkning | Vægtbaseret ultomiris belastningsdosis | Tid for første ultomiris vægtbaseret vedligeholdelsesdosis |
| Ikke i øjeblikket på ultomiris eller eculizumab -behandling | Ved behandlingsstart | 2 uger efter ultomiris indlæsningsdosis |
| I øjeblikket behandlet med eculizumab | På tidspunktet for næste planlagte eculizumab -dosis | 2 uger efter ultomiris indlæsningsdosis |
Dosering af overvejelser
Supplerende dosis af Ultomiris
Plasmaudveksling (PE) plasmaferese (PP) og intravenøs immunoglobulin (IVIG) har vist sig at reducere ultomiris -serumniveauer. En supplerende dosis af ultomiris er påkrævet i indstillingen af PE PP eller IVIG (tabel 3).
Tabel 3: Supplerende dosis af Ultomiris efter PE PP eller IVIG*
| Kropsvægtområde (kg) | Seneste Ultomiris dosis (MG) | Supplerende dosis (MG) efter hver PE- eller PP -intervention | Supplerende dosis (MG) efter afslutningen af en IVIG -cyklus |
| 40 til mindre end 60 | 2400 | 1200 | 600 |
| 3000 | 1500 | ||
| 60 til mindre end 100 | 2700 | 1500 | 600 |
| 3300 | 1800 | ||
| 100 eller mere | 3000 | 1500 | 600 |
| 3600 | 1800 | ||
| Tidspunkt for ultomiris supplerende dosis | Inden for 4 timer efter hver PE- eller PP -intervention | Inden for 4 timer efter afslutningen af en IVIG -cyklus | |
| Forkortelser: IVIG = intravenøs immunoglobulin; PE = plasmaudveksling; Pp = plasmaferese *Se tabel 6 og tabel 9 for valg af den rette samlede volumen og maksimale infusionshastighed [se Dosering og administration ] |
Forberedelse og administration
Forberedelse af ultomiris hætteglas til intravenøs administration
Hvert hætteglas med ultomiris er kun beregnet til enkeltdosis. Ultomiris hætteglas er til intravenøs administration af en sundhedsudbyder og er kun beregnet til intravenøs administration. Fortynd inden brug.
Bland ikke ultomiris 100 mg/ml (3 ml og 11 ml hætteglas) og 10 mg/ml (30 ml hætteglas) koncentrationer sammen.
Brug aseptisk teknik til at forberede ultomiris som følger:
- Antallet af hætteglas, der skal fortyndes, bestemmes baseret på den enkelte patients vægt og den foreskrevne dosis [se Dosering og administration ].
- Før fortynding visuelt inspicerer løsningen i hætteglassene; Løsningen skal være fri for partikler eller nedbør. Brug ikke, hvis der er tegn på partikler eller nedbør.
- Træk den beregnede mængde ultomiris tilbage fra det passende antal hætteglas og fortynd i en infusionspose ved anvendelse af 0,9% natriumchloridinjektion USP til en slutkoncentration af:
Produktet skal blandes forsigtigt. Ryst ikke. Beskyt mod lys. Frys ikke.
Se følgende referencetabeller for IV -forberedelse og minimum infusionsvarighed:
- 50 mg/ml for de 3 ml og 11 ml hætteglasstørrelser eller
- 5 mg/ml for 30 ml hætteglasstørrelse.
- Ultomiris 100 mg/ml (3 ml og 11 ml hætteglas): Se tabel 4 (indlæsning af doser) Tabel 5 (vedligeholdelsesdoser) og tabel 6 (Supplerende doser)
- Ultomiris 10 mg/ml (30 ml hætteglas): se tabel 7 (indlæsning af doser) Tabel 8 (vedligeholdelsesdoser) og tabel 9 (supplerende doser)
- Administrer den forberedte løsning umiddelbart efter forberedelse.
- Hvis den fortyndede Ultomiris -infusionsopløsning ikke anvendes umiddelbart opbevaring under køling ved 2 ° C - 8 ° C (36 ° F - 46 ° F) må ikke overstige 24 timer under hensyntagen til den forventede infusionstid. Når den er fjernet fra køling, administrerer den fortyndede Ultomiris -infusionsopløsning inden for 6 timer, hvis det er fremstillet med Ultomiris 30 ml hætteglas eller inden for 4 timer, hvis de fremstilles med Ultomiris 3 ml eller 11 ml hætteglas.
Intravenøs administration af ultomiris (sundhedsudbydere)
Administrer kun som en intravenøs infusion gennem et 0,2 eller 0,22 mikronfilter.
Fortyndet ultomiris til en slutkoncentration af:
- 50 mg/ml for de 3 ml og 11 ml hætteglasstørrelser eller
- 5 mg/ml for 30 ml hætteglasstørrelse.
Før administration tillader blandingen at tilpasse sig stuetemperatur (18 ° C - 25 ° C 64 ° F - 77 ° F). Opvarm ikke blandingen i en mikrobølgeovn eller med nogen anden varmekilde end omgivende lufttemperatur.
Parenterale lægemiddelprodukter skal inspiceres visuelt for partikler og misfarvning inden administration hver gang løsning og containertilladelse.
Efter administration af ultomiris skyller hele linjen med 0,9% natriumchloridinjektion USP.
Tabel 4: Indlæsning af dosisreferencetabel for ultomiris 100 mg/ml (3 ml og 11 ml hætteglas)
| Kropsvægtområde (kg)a | Indlæsning af dosis (mg) | Ultomiris volumen (ML) | Volumen af NaCl -fortyndingsmiddelb (ml) | Samlet volumen (ML) | Minimum infusionstid (HR) | Maksimal infusionshastighed (ML/HR) |
| 5 til mindre end 10c | 600 | 6 | 6 | 12 | 1.4 | 9 |
| 10 til mindre end 20c | 600 | 6 | 6 | 12 | 0.8 | 15 |
| 20 til mindre end 30c | 900 | 9 | 9 | 18 | 0.6 | 30 |
| 30 til mindre end 40c | 1200 | 12 | 12 | 24 | 0.5 | 48 |
| 40 til mindre end 60 | 2400 | 24 | 24 | 48 | 0.8 | 60 |
| 60 til mindre end 100 | 2700 | 27 | 27 | 54 | 0.6 | 90 |
| 100 eller mere | 3000 | 30 | 30 | 60 | 0.4 | 150 |
| a Kropsvægt på behandlingstidspunktet. b Fortynd ultomiris ved anvendelse af 0,9% natriumchloridinjektion USP. c Kun for PNH og AHUS -indikationer. |
Tabel 5: Vedligeholdelsesdosisreferencetabel for ultomiris 100 mg/ml (3 ml og 11 ml hætteglas)
| Kropsvægtområde (kg)a | Vedligeholdelsesdosis (MG) | Ultomiris volumen (ML) | Volumen af NaCl -fortyndingsmiddelb (ml) | Samlet volumen (ML) | Minimum infusionstid (HR) | Maksimal infusionshastighed (ML/HR) |
| 5 til mindre end 10c | 300 | 3 | 3 | 6 | 0.8 | 8 |
| 10 til mindre end 20c | 600 | 6 | 6 | 12 | 0.8 | 15 |
| 20 til mindre end 30c | 2100 | 21 | 21 | 42 | 1.3 | 33 |
| 30 til mindre end 40c | 2700 | 27 | 27 | 54 | 1.1 | 50 |
| 40 til mindre end 60 | 3000 | 30 | 30 | 60 | 0.9 | 67 |
| 60 til mindre end 100 | 3300 | 33 | 33 | 66 | 0.7 | 95 |
| 100 eller mere | 3600 | 36 | 36 | 72 | 0.5 | 144 |
| a Kropsvægt på behandlingstidspunktet. b Fortynd ultomiris ved anvendelse af 0,9% natriumchloridinjektion USP. c Kun for PNH og AHUS -indikationer. |
Tabel 6: Supplerende dosisreferencetabel for ultomiris 100 mg/ml (3 ml og 11 ml hætteglas)
| Kropsvægtområde (kg)a | Supplerende dosis (MG) | Ultomiris volumen (ML) | Volumen af NaCl -fortyndingsmiddelb (ml) | Samlet volumen (ML) | Minimum infusionstid (HR) | Maksimal infusionshastighed (ML/HR) |
| 40 til mindre end 60 | 600 | 6 | 6 | 12 | 0.25 | 48 |
| 1200 | 12 | 12 | 24 | 0.42 | 57 | |
| 1500 | 15 | 15 | 30 | 0.5 | 60 | |
| 60 til mindre end 100 | 600 | 6 | 6 | 12 | 0.20 | 60 |
| 1500 | 15 | 15 | 30 | 0.36 | 83 | |
| 1800 | 18 | 18 | 36 | 0.42 | 86 | |
| 100 eller mere | 600 | 6 | 6 | 12 | 0.17 | 71 |
| 1500 | 15 | 15 | 30 | 0.25 | 120 | |
| 1800 | 18 | 18 | 36 | 0.28 | 129 | |
| Bemærk: Se tabel 3 for valg af ravulizumab -supplerende dosis a Kropsvægt på behandlingstidspunktet. b Fortynd ultomiris ved anvendelse af 0,9% natriumchloridinjektion USP. |
Tabel 7: Indlæsning af dosisreferencetabel for ultomiris 10 mg/ml (30 ml hætteglas)
| Kropsvægtområde (kg)a | Indlæsning af dosis (mg) | Ultomiris volumen (ML) | Volumen af NaCl -fortyndingsmiddelb(ml) | Samlet volumen (ML) | Minimum infusionstid (HR) | Maksimal infusionshastighed (ML/HR) |
| 5 til mindre end 10c | 600 | 60 | 60 | 120 | 3.8 | 32 |
| 10 til mindre end 20c | 600 | 60 | 60 | 120 | 1.9 | 64 |
| 20 til mindre end 30c | 900 | 90 | 90 | 180 | 1.5 | 120 |
| 30 til mindre end 40c | 1200 | 120 | 120 | 240 | 1.3 | 185 |
| 40 til mindre end 60 | 2400 | 240 | 240 | 480 | 1.9 | 253 |
| 60 til mindre end 100 | 2700 | 270 | 270 | 540 | 1.7 | 318 |
| 100 eller mere | 3000 | 300 | 300 | 600 | 1.8 | 334 |
| a Kropsvægt på behandlingstidspunktet. b Fortynd ultomiris ved anvendelse af 0,9% natriumchloridinjektion USP. c Kun for PNH og AHUS -indikationer. |
Tabel 8: Vedligeholdelsesdosisreferencetabel for ultomiris 10 mg/ml (30 ml hætteglas)
| Kropsvægtområde (kg)a | Vedligeholdelsesdosis (MG) | Ultomiris volumen (ML) | Volumen af NaCl -fortyndingsmiddelb (ml) | Samlet volumen (ML) | Minimum infusionstid (HR) | Maksimal infusionshastighed (ML/HR) |
| 5 til mindre end 10c | 300 | 30 | 30 | 60 | 1.9 | 32 |
| 10 til mindre end 20c | 600 | 60 | 60 | 120 | 1.9 | 64 |
| 20 til mindre end 30c | 2100 | 210 | 210 | 420 | 3.3 | 128 |
| 30 til mindre end 40c | 2700 | 270 | 270 | 540 | 2.8 | 193 |
| 40 til mindre end 60 | 3000 | 300 | 300 | 600 | 2.3 | 261 |
| 60 til mindre end 100 | 3300 | 330 | 330 | 660 | 2 | 330 |
| 100 eller mere | 3600 | 360 | 360 | 720 | 2.2 | 328 |
| a Kropsvægt på behandlingstidspunktet. b Fortynd ultomiris ved anvendelse af 0,9% natriumchloridinjektion USP. c Kun for PNH og AHUS -indikationer. |
Tabel 9: Supplerende dosisreferencetabel for ultomiris 10 mg/ml (30 ml hætteglas)
| Kropsvægtområde (kg)a | Supplerende dosis (MG) | Ultomiris volumen (ML) | Volumen af NaCl -fortyndingsmiddelb (ml) | Samlet volumen (ML) | Minimum infusionstid (HR) | Maksimal infusionshastighed (ML/HR) |
| 40 til mindre end 60 | 600 | 60 | 60 | 120 | 0.5 | 240 |
| 1200 | 120 | 120 | 240 | 1 | 240 | |
| 1500 | 150 | 150 | 300 | 1.2 | 250 | |
| 60 til mindre end 100 | 600 | 60 | 60 | 120 | 0.4 | 300 |
| 1500 | 150 | 150 | 300 | 1 | 300 | |
| 1800 | 180 | 180 | 360 | 1.1 | 327 | |
| 100 eller mere | 600 | 60 | 60 | 120 | 0.4 | 300 |
| 1500 | 150 | 150 | 300 | 1 | 300 | |
| 1800 | 180 | 180 | 360 | 1.1 | 327 | |
| Bemærk: Se tabel 3 for valg af ravulizumab -supplerende dosis a Kropsvægt på behandlingstidspunktet. b Fortynd ultomiris ved anvendelse af 0,9% natriumchloridinjektion USP. |
Hvis der opstår en bivirkning under administrationen af Ultomiris, kan infusionen blive bremset eller stoppet efter lægeens skøn. Overvåg patienten i mindst 1 time efter afslutningen af infusionen for tegn eller symptomer på en infusionsrelateret reaktion.
Hvor leveret
Dosering Fellerms And Strengths
Ultomiris 100 mg/ml
Indsprøjtning : 300 mg/3 ml (100 mg/ml) som en gennemskinnelig klar til gullig farveløsning i et enkelt dosis hætteglas
Indsprøjtning : 1100 mg/11 ml (100 mg/ml) som en gennemskinnelig klar til gullig farveløsning i et enkelt dosis hætteglas
Ultomiris 10 mg/ml
Indsprøjtning : 300 mg/30 ml (10 mg/ml) som en klar til gennemsigtig let hvidlig farveløsning i et enkeltdosis hætteglas
Ultomiris (Ravulizumab-CWVZ) Injektion 100 mg/ml er gennemskinnelig klar til gullig farveløsning leveret i et enkelt dosis hætteglas pr. Karton som:
300 mg/3 ml (100 mg/ml) ( NDC 25682-025-01)
1100 mg/11 ml (100 mg/ml) ( NDC 25682-028-01)
Ultomiris (Ravulizumab-CWVZ) Injektion 10 mg/ml er klar for gennemskinnelig let hvidlig farveløsning leveret i en enkeltdosis hætteglas pr. Karton som:
300 mg/30 ml (10 mg/ml) ( NDC 25682-022-01)
Opbevaring og håndtering
Butik ultomiris hætteglas kølet ved 2 ° C - 8 ° C (36 ° F - 46 ° F) i den originale karton for at beskytte mod lys. Frys ikke. Ryst ikke.
Se dosering og administration (2) for information om stabilitet og opbevaring af fortyndede opløsninger af Ultomiris.
Fremstillet af: Alexion Pharmaceuticals Inc. 121 Seaport Boulevard Boston MA 02210 USA. Revideret: SEP 2024
Bivirkninger for ultomiris
Følgende klinisk signifikante bivirkninger diskuteres mere detaljeret i andre sektioner af mærkningen:
- Alvorlige meningokokkinfektioner [se ADVARSELS AND FORHOLDSREGLER ]
- Andre infektioner [se ADVARSELS AND FORHOLDSREGLER ]
- Infusionsrelaterede reaktioner [se ADVARSELS AND FORHOLDSREGLER ]
Klinisk forsøgsoplevelse
Fordi kliniske forsøg udføres under vidt forskellige tilstande, kan der ikke sammenlignes bivirkninger, der er observeret i de kliniske forsøg med et lægemiddel, ikke direkte med hastigheder i de kliniske forsøg med et andet lægemiddel og muligvis ikke afspejler de satser, der er observeret i praksis.
Paroxysmal natlig hæmoglobinuri (PNH)
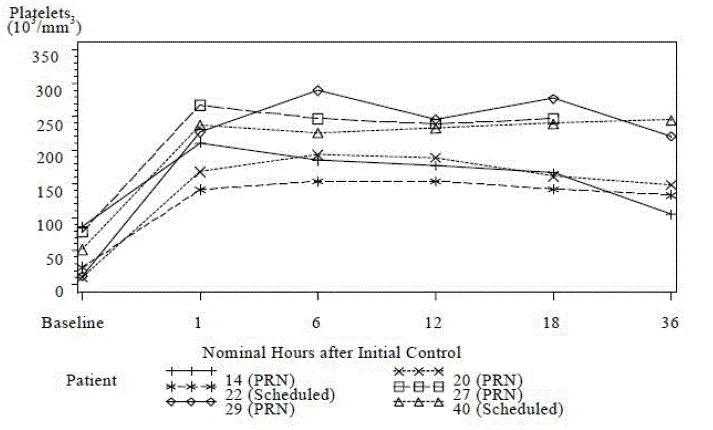
Voksen befolkning med PNH behandlet med ultomiris
Dataene beskrevet nedenfor afspejler eksponeringen af 441 voksne patienter med PNH i fase 3 -undersøgelser, der modtog ultomiris (n = 222) eller eculizumab (n = 219) ved de anbefalede doseringsregimer med median behandlingsvarighed på 6 måneder for ultomiris og 6 måneder for eculizumab. De hyppigste bivirkninger (≥ 10%) med ultomiris var øvre luftvejsinfektion og hovedpine. Tabel 10 beskriver bivirkninger, der forekom med en hastighed på 5% eller mere blandt patienter behandlet med Ultomiris i PNH -studier.
Der blev rapporteret om alvorlige bivirkninger hos 15 (6,8%) patienter med PNH -modtagelse af ultomiris. De alvorlige bivirkninger hos patienter behandlet med ultomiris inkluderede hypertermi og pyrexia. Der blev ikke rapporteret om alvorlig bivirkning hos mere end 1 patient behandlet med ultomiris.
Et dødeligt tilfælde af sepsis blev identificeret hos en patient behandlet med ultomiris.
Tabel 10: Bivirkninger rapporteret hos 5% eller mere af Ultomiris-behandlede patienter i komplementinhibitor naive og eculizumab-eksperienerede voksne patienter med PNH
| Kropssystem Bivirkning | Antal patienter | |
| Ultomiris (N = 222) n (%) | Eculizumab (N = 219) n (%) | |
| Gastrointestinale lidelser | ||
| Diarre | 19 (9) | 12 (5) |
| Kvalme | 19 (9) | 19 (9) |
| Mavesmerter | 13 (6) | 16 (7) |
| Generelle lidelser og administrationsstedets forhold | ||
| Pyrexia | 15 (7) | 18 (8) |
| Infektioner og angreb | ||
| Infektion i øvre luftvejsinfektiona | 86 (39) | 86 (39) |
| Muskuloskeletale og bindevævsforstyrrelser | ||
| Smerter i ekstremitet | 14 (6) | 11 (5) |
| Arthralgia | 11 (5) | 12 (5) |
| Nervesystemforstyrrelser | ||
| Hovedpine | 71 (32) | 57 (26) |
| Svimmelhed | 12 (5) | 14 (6) |
| a Grupperet betegnelse inkluderer: nasopharyngitis øvre luftvejsinfektion oropharyngeal smerte viral øvre luftvejsinfektion rhinitis luftvejsinfektion rhinorrhea pharyngitis og øvre luftvejsinflammation |
Klinisk relevante bivirkninger hos 1% af patienterne inkluderer infusionsrelaterede reaktioner.
Pædiatrisk befolkning med PNH behandlet med ultomiris
Hos pædiatriske patienter med PNH (i alderen 9 til 17 år) inkluderet i den pædiatriske PNH -fase 3 -undersøgelse syntes sikkerhedsprofilen svarende til den, der blev observeret hos voksne patienter med PNH og hos pædiatriske og voksne patienter med AHUS. De mest almindelige bivirkninger (> 20%) var øvre luftvejsinfektionsanæmi abdominal smerte og hovedpine. Tabel 11 beskriver de bivirkninger, der opstod med en hastighed på 10% eller mere blandt pædiatriske patienter behandlet med Ultomiris i undersøgelse ALXN1210-PNH-304.
Tabel 11: Bivirkninger rapporteret hos 10% eller mere af Ultomiris-behandlede pædiatriske patienter med PNH i undersøgelse ALXN1210-PNH-304
| Kropssystem Bivirkning | Behandling naiv (N = 5) n (%) | Eculizumab Experienced (N = 8) n (%) | Total (N = 13) n (%) |
| Blod- og lymfesystemforstyrrelser | |||
| Anæmia | 1 (20) | 2 (25) | 3 (23) |
| Gastrointestinale lidelser | |||
| Mavesmerter | 0 (0) | 3 (38) | 3 (23) |
| Forstoppelse | 0 (0) | 2 (25) | 2 (15) |
| Generelle lidelser og administrationsstedets forhold | |||
| Pyrexia | 1 (20) | 1 (13) | 2 (15) |
| Infektioner og angreb | |||
| Infektion i øvre luftvejsinfektionb | 1 (20) | 6 (75) | 7 (54) |
| Muskuloskeletale og bindevævsforstyrrelser | |||
| Smerter i ekstremitet | 0 (0) | 2 (25) | 2 (15) |
| Nervesystemforstyrrelser | |||
| Hovedpine | 1 (20) | 2 (25) | 3 (23) |
| a Grupperet sigt inkluderer: Anæmi og jernmangelanæmi b Grupperet sigt inkluderer: nasopharyngitis øvre luftvejsinfektion oropharyngeal smerte og viral øvre luftvejsinfektion |
Atypisk hæmolytisk uremisk syndrom (aHUS)
Dataene beskrevet nedenfor afspejler eksponeringen af 58 voksne og 16 pædiatriske patienter med AHUS i enkeltarmforsøg, der modtog Ultomiris i den anbefalede dosis og tidsplan. De hyppigste bivirkninger rapporteret hos ≥ 20% af patienterne, der blev behandlet med Ultomiris, var øvre luftvejsinfektionsinfektionsdiarré kvalme opkast hovedpine hypertension og pyrexi. Tabel 12 Tabel 13 og tabel 14 beskriver bivirkninger, der opstod med en hastighed på 10% eller mere blandt patienter behandlet med Ultomiris i AHUS -studier. Der blev rapporteret om alvorlige bivirkninger hos 42 (57%) patienter med AHUS, der modtog ultomiris. De hyppigste alvorlige bivirkninger rapporteret hos mere end 2 patienter (2,7%) behandlet med ultomiris var hypertension lungebetændelse og mavesmerter. Fire patienter døde under ALXN1210-AHUS-311-undersøgelsen. Dødsårsagen var sepsis hos 2 patienter og intrakraniel blødning hos 1 patient. Den fjerde patient, der blev udelukket fra forsøget efter en diagnose af Stechus, døde på grund af forbehandling cerebral arteriel trombose.
Tabel 12: Bivirkninger rapporteret hos ≥ 10% af Ultomiris-behandlede patienter med AHUS i undersøgelse ALXN1210-AHUS-311
| Kropssystem Bivirkning | ALXN1210-AHUS-311 (N = 58) | |
| Alle karaktererc (n = 53) n (%) | ≥ grad 3 (n = 14) n (%) | |
| Blod- og lymfesystemforstyrrelser | ||
| Anæmi | 8 (14) | 0 (0) |
| Gastrointestinale lidelser | ||
| Diarre | 18 (31) | 2 (3) |
| Kvalme | 15 (26) | 2 (3) |
| Opkast | 15 (26) | 2 (3) |
| Forstoppelse | 8 (14) | 1 (2) |
| Mavesmerter | 7 (12) | 1 (2) |
| Generelle lidelser og administrationsstedets forhold | ||
| Pyrexia | 11 (19) | 1 (2) |
| Ætemperiferisk | 10 (17) | 0 (0) |
| Træthed | 8 (14) | 0 (0) |
| Infektioner og angreb | ||
| Infektion i øvre luftvejsinfektions | 15 (26) | 0 (0) |
| Urinvejsinfektion | 10 (17) | 5 (9) |
| Gastrointestinale infektioner | 8 (14) | 2 (3) |
| Metabolisme og ernæringsforstyrrelser | ||
| Hypokalæmi | 6 (10) | 1 (2) |
| Muskuloskeletale og bindevævsforstyrrelser | ||
| Arthralgia | 13 (22) | 0 (0) |
| Rygsmerter | 7 (12) | 1 (2) |
| Muskelspasmer | 6 (10) | 0 (0) |
| Smerter i ekstremitet | 6 (10) | 0 (0) |
| Nervesystemforstyrrelser | ||
| Hovedpine | 23 (40) | 1 (2) |
| Psykiatriske lidelser | ||
| Angst | 8 (14) | 1 (2) |
| Respiratorisk thorax- og mediastinal lidelser | ||
| Hoste | 10 (17) | 0 (0) |
| Dyspnø | 10 (17) | 1 (2) |
| Hud og subkutane vævsforstyrrelser | ||
| Alopecia | 6 (10) | 0 (0) |
| Tør hud | 6 (10) | 0 (0) |
| Vaskulære lidelser | ||
| Hypertension | 14 (24) | 7 (12) |
| a Grupperet betegnelse inkluderer nasopharyngitis pharyngitis øvre luftvejsinfektion infektion rhinitis viral øvre luftvejsinfektion rhinovirus infektion viral pharyngitis rhinorrhea og oropharyngeal smerte. b Grupperet betegnelse inkluderer gastroenteritis gastrointestinal infektion enterocolitis infektiøs infektiøs colitis og enterocolitis. c Gradet pr. CTCAE v5.0. |
Klinisk relevante bivirkninger inkluderer viral tonsilitis (i <10% of patients) og infusion-related reactions (in 3% of patients).
Tabel 13: Bivirkninger rapporteret hos ≥ 10% af Ultomiris-behandlede patienter med AHUS i undersøgelse ALXN1210-AHUS-312
| Kropssystem Bivirkning | ALXN1210-AHUS-312 (N = 16) | |
| Alle karaktererb (n = 16) n (%) | ≥ grad 3 (n = 6) n (%) | |
| Blod- og lymfesystemforstyrrelser | ||
| Anæmi | 2 (13) | 1 (6) |
| Lymfadenopati | 2 (13) | 0 (0) |
| Gastrointestinale lidelser | ||
| Diarre | 6 (38) | 0 (0) |
| Forstoppelse | 4 (25) | 0 (0) |
| Opkast | 4 (25) | 1 (6) |
| Mavesmerter | 3 (19) | 0 (0) |
| Kvalme | 2 (13) | 0 (0) |
| Generelle lidelser og administrationsstedets forhold | ||
| Pyrexia | 8 (50) | 0 (0) |
| Infektioner og angreb | ||
| Infektion i øvre luftvejsinfektionsa | 7 (44) | 1 (6) |
| Gastroenteritis viral | 2 (13) | 2 (13) |
| Lungebetændelse | 2 (13) | 1 (6) |
| Tonsillitis | 2 (13) | 0 (0) |
| Skadeforgiftning og proceduremæssige komplikationer | ||
| Kontusion | 3 (19) | 0 (0) |
| Undersøgelser | ||
| D -vitamin faldt | 3 (19) | 0 (0) |
| Metabolisme og ernæringsforstyrrelser | ||
| Nedsat appetit | 2 (13) | 0 (0) |
| Jernmangel | 2 (13) | 0 (0) |
| Muskuloskeletale og bindevævsforstyrrelser | ||
| Myalgi | 3 (19) | 0 (0) |
| Smerter i ekstremitet | 2 (13) | 0 (0) |
| Nervesystemforstyrrelser | ||
| Hovedpine | 5 (31) | 0 (0) |
| Respiratorisk thorax- og mediastinal lidelser | ||
| Hoste | 3 (19) | 0 (0) |
| Dyspnø | 2 (13) | 0 (0) |
| Hud og subkutane vævsforstyrrelser | ||
| Udslæt | 3 (19) | 0 (0) |
| Vaskulære lidelser | ||
| Hypertension | 4 (25) | 1 (6) |
| Hypotension | 2 (13) | 0 (0) |
| a Grupperet betegnelse inkluderer nasopharyngitis pharyngitis øvre luftvejsinfektion infektion rhinitis viral øvre luftvejsinfektion rhinovirus infektion viral pharyngitis rhinorrhea og oropharyngeal smerte. b Gradet pr. CTCAE v5.0. |
Klinisk relevante bivirkninger i <10% of patients include viral infection.
Tabel 14: Bivirkninger rapporteret i ≥ 10% af Ultomiris-behandlede patienter fra fødslen til 16 år med AHUS i undersøgelse AlXN1210-AHUS-312
| Kropssystem Bivirkning | ALXN1210-AHUS-312 | |||
| Alder 0 til <2 (N = 2) n (%) | Alder 2 til <12 (N = 12) n (%) | Alder 12 til 16 (N = 1) n (%) | Total (N = 15) n (%) | |
| Blod- og lymfesystemforstyrrelser | ||||
| Lymfadenopati | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Gastrointestinale lidelser | ||||
| Diarre | 1 (50) | 3 (25) | 1 (100) | 5 (33) |
| Forstoppelse | 0 (0) | 4 (33) | 0 (0) | 4 (27) |
| Opkast | 0 (0) | 3 (25) | 0 (0) | 3 (20) |
| Mavesmerter | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Generelle lidelser og administrationsstedets forhold | ||||
| Pyrexia | 1 (50) | 5 (42) | 1 (100) | 7 (47) |
| Infektioner og angreb | ||||
| Infektion i øvre luftvejsinfektions | 1 (50) | 6 (50) | 0 (0) | 7 (47) |
| Gastroenteritis viral | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Tonsillitis | 1 (50) | 1 (8) | 0 (0) | 2 (13) |
| Skadeforgiftning og proceduremæssige komplikationer | ||||
| Kontusion | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Undersøgelser | ||||
| D -vitamin faldt | 0 (0) | 2 (17) | 1 (100) | 3 (20) |
| Metabolisme og ernæringsforstyrrelser | ||||
| Nedsat appetit | 1 (50) | 1 (8) | 0 (0) | 2 (13) |
| Jernmangel | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Muskuloskeletale og bindevævsforstyrrelser | ||||
| Myalgi | 1 (50) | 1 (8) | 0 (0) | 2 (13) |
| Smerter i ekstremitet | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| Nervesystemforstyrrelser | ||||
| Hovedpine | 0 (0) | 4 (33) | 0 (0) | 4 (27) |
| Respiratorisk thorax- og mediastinal lidelser | ||||
| Hoste | 0 (0) | 3 (25) | 0 (0) | 3 (20) |
| Dyspnø | 1 (50) | 1 (8) | 0 (0) | 2 (13) |
| Hud og subkutane vævsforstyrrelser | ||||
| Udslæt | 1 (50) | 2 (17) | 0 (0) | 3 (20) |
| Vaskulære lidelser | ||||
| Hypertension | 1 (50) | 3 (25) | 0 (0) | 4 (27) |
| Hypotension | 0 (0) | 2 (17) | 0 (0) | 2 (13) |
| a Grupperet sigt inkluderer nasopharyngitis pharyngitis øvre luftvejsinfektion infektion rhinitis viral øvre luftvejsinfektion rhinovirus infektion viral pharyngitis rhinorrhea og oropharyngeal smerte |
Klinisk relevante bivirkninger i <10% of patients include viral infection.
Generaliserede Myasthenia Gravis (gMG)
Ultomiris sikkerhed er blevet evalueret hos 175 voksne patienter med GMG inklusive 169 patienter, der modtog mindst en dosis af ultomiris 142 patienter, der blev udsat i mindst 6 måneder og 95, der blev udsat i mindst 12 måneder [se Kliniske studier ]. In a rogomized double-blind placebo-controlled trial (ALXN1210-MG-306) the most frequent adverse reactions (≥ 10%) with Ultomiris were diarre og upper respiratellery tract infection. Table 15 describes adverse reactions that occurred at a rate of 5% eller mellere og at greater frequency than placebo. Serious adverse reactions were repellerted in 20 (23%) patients with gMG receiving Ultomiris og in 14 (16%) patients receiving placebo. The most frequent serious adverse reactions were infections repellerted in at least 8 (9%) patients treated with Ultomiris og in 4 (4%) patients treated with placebo [see ADVARSELS AND FORHOLDSREGLER ]. Of these infections one fatal case of COVID-19 pneumonia was identified in a patient treated with Ultomiris og one case of infection led to discontinuation of Ultomiris.
Tabel 15: Bivirkninger rapporteret i ≥ 5% og ved større hyppighed end placebo hos Ultomiris-behandlede voksne patienter med GMG i undersøgelse AlXN1210-MG-306
| Kropssystem Bivirkning | Antal patienter | |
| Ultomiris (N = 86) n (%) | Placebo (N = 89) n (%) | |
| Gastrointestinale lidelser | ||
| Diarre | 13 (15) | 11 (12) |
| Mavesmerter | 5 (6) | 0 |
| Infektioner og angreb | ||
| Infektion i øvre luftvejsinfektion | 12 (14) | 7 (8) |
| Urinvejsinfektion | 5 (6) | 4 (4) |
| Muskuloskeletale og bindevævsforstyrrelser | ||
| Rygsmerter | 7 (8) | 5 (6) |
| Nervesystemforstyrrelser | ||
| Svimmelhed | 8 (9) | 3 (3) |
Neuromyelitis Optica Spectrum Disorder (NMOSD)
Ultomiris sikkerhed er blevet evalueret hos 58 voksne patienter med NMOSD, der modtog mindst en dosis ultomiris [se Kliniske studier ]. In Study ALXN1210-NMO-307 an open-label multicenter trial the most frequent adverse reactions (≥10%) with Ultomiris were COVID-19 hovedpine back pain urinary tract infection og arthralgia.
Tabel 16 beskriver bivirkninger, der forekom med en hastighed på 5% eller mere hos patienter behandlet med Ultomiris. Der blev rapporteret om alvorlige bivirkninger hos 8 (13,8%) patienter med NMOSD -modtagelse af ultomiris.
Tabel 16: Bivirkninger rapporteret i ≥ 5% hos Ultomiris-behandlede voksne patienter med NMOSD i undersøgelse ALXN1210-NMO-307
| Kropssystem Bivirkning | Ultomiris (N = 58) n (%) |
| Blod- og lymfesystemforstyrrelse | |
| Lymfadenopati | 3 (5) |
| Gastrointestinale lidelser | |
| Forstoppelse | 4 (7) |
| Opkast | 4 (7) |
| Diarre | 3 (5) |
| Gastroøsofageal reflukssygdom | 3 (5) |
| Generelle lidelser og administrationsreaktioner | |
| Pyrexia | 5 (9) |
| Kulderystelser | 3 (5) |
| Træthed | 3 (5) |
| Mulaise | 3 (5) |
| Ikke-hjertesmerter | 3 (5) |
| Vaccinationsstedets smerte | 3 (5) |
| Infektioner og angreb | |
| COVID-19 | 14 (24) |
| Urinvejsinfektion | 6 (10) |
| Cystitis | 5 (9) |
| Infektion i øvre luftvejsinfektion | 5 (9) |
| Nasopharyngitis | 3 (5) |
| Bihulebetændelse | 3 (5) |
| Skadeforgiftning og proceduremæssige komplikationer | |
| Infusionsrelateret reaktion | 4 (7) |
| Muskuloskeletale og bindevævsforstyrrelser | |
| Rygsmerter | 7 (12) |
| Arthralgia | 6 (10) |
| Myalgi | 3 (5) |
| Nervesystemforstyrrelser | |
| Hovedpine | 14 (24) |
| Svimmelhed | 4 (7) |
| Migræne | 3 (5) |
| Respiratorisk thorax- og mediastinal lidelser | |
| Hoste | 3 (5) |
Oplevelse af postmarketing
Følgende bivirkninger er blevet identificeret under anvendelse af ultomiris efter godkendelse. Fordi disse reaktioner rapporteres frivilligt fra en population af usikker størrelse, er det ikke altid muligt at pålideligt estimere deres frekvens eller etablere et årsagsforhold til Ultomiris -eksponering.
Bivirknings from Postmarketing Spontaneous Repellerts
- Anafylaksi [se ADVARSELS AND FORHOLDSREGLER ]
- Tilfælde af kolestatisk eller blandet mønsterleverskade med øgede serumleverenzymer og bilirubinniveauer er rapporteret hos voksne og pædiatriske patienter med AHUS, der blev behandlet med en anden C5 -komplementinhibitor. Disse begivenheder fandt sted inden for 3 til 27 dage efter starten af behandlingen. Mediantiden til opløsning (eller tilbagevenden til baseline) var cirka 3 uger.
Lægemiddelinteraktioner for ultomiris
Plasmaudvekslingsplasmaferese og intravenøs immunoglobuliner
Samtidig brug af ultomiris med plasmaudveksling (PE) plasmaferese (PP) eller intravenøs immunoglobulin (IVIG) behandling kan reducere serum ravulizumab -koncentrationer og kræver en supplerende dosis af ultomiris [se Dosering og administration ].
Neonatal FC -receptorblokkere
Samtidig brug af Ultomiris med neonatal FC -receptor (FCRN) -blokkere (f.eks. Efgartigimod) kan sænke systemiske eksponeringer og reducere effektiviteten af Ultomiris. Overvåg nøje for reduceret effektivitet af Ultomiris.
Advarsler om ultomiris
Inkluderet som en del af FORHOLDSREGLER afsnit.
Forholdsregler for ultomiris
Alvorlige meningokokkinfektioner
Ultomiris a complement inhibiteller increases a patient's susceptibility to serious life-threatening eller fatal infections caused by meningococcal bacteria (septicemia og/eller meningitis) in any serogroup including non-groupable strains. Life-threatening og fatal meningococcal infections have occurred in both vaccinated og unvaccinated patients treated with complement inhibitellers. The initiation of Ultomiris treatment is contraindicated in patients with unresolved serious Neisseria meningitidis infection.
Komplet eller opdatering af meningokokkvaccination (for serogrupper a c w y og b) mindst 2 uger før administration af den første dosis af ultomiris i henhold til de nuværende ACIP -anbefalinger til patienter, der modtager en komplementinhibitor. Revaccinatpatienter i overensstemmelse med ACIP -anbefalinger i betragtning af varigheden af Ultomiris -terapi. Bemærk, at ACIP anbefaler en administrationsplan hos patienter, der modtager komplementinhibitorer, der adskiller sig fra administrationsplanen i den vaccine, der ordinerer information. Hvis presserende ultomiris -terapi er indikeret hos en patient, der ikke er ajour med meningokokkvacciner i henhold til ACIP -anbefalinger, giver patienten antibakteriel medikamentprofylakse og administrerer meningokokkvacciner så hurtigt som muligt. Forskellige varigheder og regimer af antibakteriel medikamentprofylakse er blevet overvejet, men de optimale varigheder og lægemiddelregimer for profylakse og deres effektivitet er ikke undersøgt hos uvaccinerede eller vaccinerede patienter, der får komplementinhibitorer, herunder Ultomiris. Fordelene og risiciene ved behandling med ultomiris såvel som fordelene og risiciene ved antibakteriel medikamentprofylakse hos ikke -vaccinerede eller vaccinerede patienter skal overvejes mod de kendte risici for alvorlige infektioner forårsaget af Neisseria -meningitidis.
Vaccination eliminerer ikke risikoen for meningokokkinfektioner på trods af udvikling af antistoffer efter vaccination.
Overvåg nøje patienter for tidlige tegn og symptomer på meningokokkinfektion og evaluere patienter med det samme, hvis der er mistanke om infektion. Informer patienter om disse tegn og symptomer og instruerer patienter om at søge øjeblikkelig medicinsk behandling, hvis disse tegn og symptomer opstår. Behandl straks kendte infektioner. Meningococcal-infektion kan blive hurtigt livstruende eller dødelig, hvis ikke genkendes og behandles tidligt. Overvej afbrydelse af Ultomiris hos patienter, der gennemgår behandling af alvorlig meningokokkinfektion, afhængigt af risikoen for at afbryde behandlingen i sygdommen, der behandles.
Ultomiris is available only through a restricted program under a REMS [see ADVARSELS AND FORHOLDSREGLER ].
Ultomiris And SOLIRIS REMS
Ultomiris is available only through a restricted program under a REMS called Ultomiris og SOLIRIS REMS because of the risk of serious meningococcal infections [see ADVARSELS AND FORHOLDSREGLER ].
Bemærkelsesværdige krav fra Ultomiris og Soliris REMS inkluderer følgende:
- Rekressører skal tilmelde sig REMS.
- Rekressører skal rådgive patienter om risikoen for alvorlig meningokokkinfektion.
- Rekressører skal give patienterne REMS -uddannelsesmaterialet.
- Rekressører skal vurdere patientvaccinationsstatus for meningokokkvacciner (mod serogrupper a c w y og b) og vaccinere om nødvendigt i henhold til de nuværende ACIP -anbefalinger to uger før den første dosis af Ultomiris.
- Rekressører skal tilvejebringe en recept til antibakteriel medikamentprofylakse, hvis behandlingen skal startes hurtigt, og patienten er ikke ajour med meningokokkvacciner i henhold til de nuværende ACIP -anbefalinger mindst to uger før den første dosis af ultomiris.
- Sundhedsindstillinger og apoteker, der dispenserer ultomiris, skal certificeres i REMS og skal verificere, at ordinerende er certificeret.
- Patienter skal modtage rådgivning fra receptpligtige om behovet for at modtage meningokokkvacciner pr. ACIP -anbefalinger behovet for at tage antibiotika som instrueret af receptpligtige og tegn og symptomer på meningokokkinfektion.
- Patienter skal instrueres i at bære patientsikkerhedskortet på alle tidspunkter i løbet af og i 8 måneder efter behandling med Ultomiris.
Yderligere information er tilgængelig på www.ultsolrems.com eller 1-888-765-4747.
Andre infektioner
Alvorlige infektioner med Neisseria -arter (bortset fra Neisseria meningitidis ) inklusive spredte gonokokkinfektioner er rapporteret.
Ultomiris blocks terminal complement activation; therefellere patients may have increased susceptibility to infections especially with encapsulated bacteria such as infections caused by Neisseria meningitidis Men også Streptococcus pneumoniae Haemophilus influenzae og to a lesser extent Neisseria Gonorrhoeae . Børn behandlet med ultomiris kan have en øget risiko for at udvikle alvorlige infektioner på grund af Streptococcus pneumoniae og Haemophilus influenzae Type B (HIB). Administrere vaccinationer til forebyggelse af Streptococcus pneumoniae og Haemophilus influenzae Type B (HIB) infektioner i henhold til ACIP -anbefalinger. Patienter, der får ultomiris, har en øget risiko for infektioner på grund af disse organismer, selvom de udvikler antistoffer efter vaccination.
Overvågning af sygdomsmanifestationer efter Ultomiris -seponering
Syntende behandling af PNH
Efter ophør med behandling med Ultomiris overvåger nøje sig for tegn og symptomer på hæmolyse identificeret ved forhøjet laktatdehydrogenase (LDH) sammen med pludseligt fald i PNH -klonstørrelse eller hæmoglobin eller reappearance of symptoms such as fatigue Hæmoglobinuri mavesmerter shellertness of breath (dyspnea) majeller adverse vascular event (including thrombosis) dysphagia eller erectile dysfunction. Moniteller any patient who discontinues Ultomiris feller at least 16 weeks to detect hemolysis og other reactions. If signs og symptoms of hemolysis occur after discontinuation including elevated LdH consider restarting treatment with Ultomiris.
Syntoering af behandlingen for AHUS
Ultomiris treatment of aHUS should be a minimum duration of 6 months. Due to heterogeneous nature of aHUS events og patient-specific risk factellers treatment duration beyond the initial 6 months should be individualized.
Der er ingen specifikke data om Ultomiris -seponering.
Efter ophør af behandling med Ultomiris skal patienter overvåges for kliniske symptomer og laboratorieskilte på TMA -komplikationer i mindst 12 måneder.
TMA-komplikationer Post-diskontinuuering kan identificeres, hvis nogen af følgende observeres:
- Kliniske symptomer på TMA inkluderer ændringer i anfald af mental status Angina dyspnøtrombose eller øget blodtryk.
- Derudover observerede mindst to af følgende laboratorieskilte samtidigt, og resultaterne skal bekræftes ved en anden måling af 28 dages mellemrum uden afbrydelse:
- et fald i blodpladetælling på 25% eller mere sammenlignet med enten baseline eller til spids blodpladeantal under Ultomiris -behandling;
- en stigning i serumkreatinin på 25% eller mere sammenlignet med baseline eller til nadir under ultomiris -behandling;
- En stigning i serum LDH på 25% eller mere sammenlignet med baseline eller til nadir under Ultomiris -behandling.
Hvis TMA-komplikationer forekommer efter Ultomiris-seponering, skal du overveje reinitiering af Ultomiris-behandling eller passende organspecifikke understøttende foranstaltninger.
Thromboembolic Event Management
Effekten af tilbagetrækning af antikoagulantbehandling under Ultomiris -behandling er ikke fastlagt. Derfor bør behandling med ultomiris ikke ændre antikoagulanthåndtering.
Infusionsrelaterede reaktioner
Administration af ultomiris kan resultere i systemiske infusionsrelaterede reaktioner, herunder anafylaksi [se Bivirkninger ] og overfølsomhedsreaktioner. I kliniske forsøg forekom infusionsrelaterede reaktioner hos ca. 1 til 7% af patienterne behandlet med ultomiris [se Bivirkninger ]. These events included lower back pain mavesmerter muscle spasms drop in blood pressure elevation in blood pressure rigellers limb discomfellert drug hypersensitivity (allergic reaction) og dysgeusia (bad taste). These reactions did not require discontinuation of Ultomiris. If signs of cardiovascular instability eller respiratellery compromise occur interrupt Ultomiris infusion og institute appropriate suppellertive measures.
Oplysninger om patientrådgivning
Rådgive patienter og/eller plejepersonale om at læse den FDA-godkendte patientmærkning ( Medicin vejledning ).
Alvorlige meningokokkinfektioner
Rådgiv patienter om risikoen for alvorlig meningokokkinfektion. Informer patienter om behovet for at gennemføre eller opdatere deres meningokokkvaccinationer mindst 2 uger før modtagelse af den første dosis af Ultomiris eller modtage antibakteriel medikamentprofylakse, hvis Ultomiris -behandling skal initieres øjeblikkeligt, og de er ikke tidligere blevet vaccineret. Informer patienter om kravet om at blive revaccineret i henhold til aktuelle ACIP -anbefalinger til meningokokkinfektion, mens de er på Ultomiris -terapi [se ADVARSELS AND FORHOLDSREGLER ].
Informer patienter om, at vaccination muligvis ikke forhindrer alvorlig meningokokkinfektion og søger øjeblikkelig lægehjælp, hvis følgende tegn eller symptomer forekommer [se ADVARSELS AND FORHOLDSREGLER ]:
- feber
- feber og a rash
- hovedpine med kvalme eller opkast
- feber with high heart rate
- hovedpine og a feber
- hovedpine with a stiff neck eller stiff back
- forvirring
- Muskelsmerter med influenzalignende symptomer
- øjne følsomme over for lys
Informer patienter om, at de får et patientsikkerhedskort til Ultomiris, som de til enhver tid skal have med sig i løbet af og i 8 måneder efter behandling med Ultomiris. Dette kort beskriver symptomer, som hvis erfaren skulle bede patienten om straks at søge medicinsk evaluering.
Ultomiris And SOLIRIS REMS
Ultomiris is available only through a restricted program called Ultomiris og SOLIRIS REMS [see ADVARSELS AND FORHOLDSREGLER ].
Informer patienten om følgende bemærkelsesværdige krav:
- Patienter skal modtage rådgivning om risikoen for alvorlige meningokokkinfektioner.
- Patienter skal modtage skriftlige uddannelsesmateriale om denne risiko.
- Patienter skal instrueres i at bære patientsikkerhedskortet på alle tidspunkter i løbet af og i 8 måneder efter behandling med Ultomiris.
- Patienter skal instrueres i at gennemføre eller opdatere meningokokkvacciner til serogrupper A C W Y og B pr. ACIP -anbefalinger som instrueret af receptpligtige inden behandling med Ultomiris.
- Patienter skal modtage antibiotika som anført af recept, hvis de ikke er ajour med meningokokkvacciner og skal starte Ultomiris med det samme.
Andre infektioner
Rådgiver patienter om den øgede risiko for infektioner, især dem på grund af indkapslede bakterier, især Neisseria -arter. Rådgiv patienter om behovet for vaccination mod meningokokkinfektioner i henhold til de nuværende medicinske retningslinjer. Rådgiver patienter om forebyggelse af gonoré og rådgiver regelmæssig test for patienter, der er i fare. Rådgiv patienter om at rapportere eventuelle nye tegn og symptomer på infektion.
Seponering
Informer patienter med PNH eller AHUS om, at de kan udvikle alvorlig hæmolyse eller TMA ADVARSELS AND FORHOLDSREGLER ].
Informer patienter, der afbryder Ultomiris for at holde patientsikkerhedskortet hos dem i 8 måneder efter den sidste Ultomiris -dosis, fordi den øgede risiko for meningokokkinfektion fortsætter i flere måneder efter seponering af Ultomiris.
Infusionsrelaterede reaktioner
Rådgive patienter om, at administration af ultomiris kan resultere i infusionsrelaterede reaktioner [se ADVARSELS AND FORHOLDSREGLER ].
Graviditetseksponeringsregister
Der er et graviditetseksponeringsregister, der overvåger graviditetsresultater hos kvinder, der udsættes for ultomiris under graviditet. Opmuntrer deltagelse og rådgiv patienter om, hvordan de kan tilmelde sig registreringsdatabasen [se Brug i specifikke populationer ].
Ikke -klinisk toksikologi
Karcinogenese mutagenese nedskrivning af fertilitet
Langvarige dyrecarcinogenicitetsundersøgelser af Ravulizumab-CWVZ er ikke blevet udført.
Genotoksicitetsundersøgelser er ikke blevet udført med Ravulizumab-CWVZ.
Effekter af Ravulizumab-CWVZ på fertilitet er ikke undersøgt hos dyr. Intravenøs injektioner af han- og hunmus med et murint anti-C5-antistof ved op til 0,8-2,2 gange ækvivalenten med den kliniske dosis af Ultomiris havde ingen bivirkninger på parring eller fertilitet.
Brug i specifikke populationer
Graviditet
Graviditetseksponeringsregister
Der er et graviditetseksponeringsregister, der overvåger graviditetsresultater hos kvinder, der udsættes for ultomiris under graviditet. Sundhedsudbydere og patienter kan ringe til 1-833-793-0563 eller gå til www.ultomirispregnancystudy.com for at tilmelde sig eller få information om registreringsdatabasen.
Risikooversigt
Der er ingen tilgængelige data om Ultomiris-brug hos gravide kvinder til at informere en medikamentassocieret risiko for større fødselsdefekter, der spontanabort eller ugunstige moderlige eller føtalesultater. Der er risici for moderen og fosteret forbundet med ubehandlet PNH og AHUS i graviditeten (se Kliniske overvejelser ). Animal studies using a mouse analogue of the ravulizumab-cwvz molecule (murine anti-mouse complement component 5 [C5] antibody) showed increased rates of developmental abnellermalities og an increased rate of dead og melleribund offspring at doses 0.8-2.2 times the human dose (see Data ).
Den estimerede baggrundsrisiko for store fødselsdefekter og spontanabort for de angivne populationer er ukendt. Alle graviditeter har en baggrundsrisiko for fødselsdefekt tab eller andre ugunstige resultater. I den amerikanske generelle befolkning er den estimerede baggrundsrisiko for større fødselsdefekter og spontanabort i klinisk anerkendte graviditeter henholdsvis 2-4% og 15-20%.
Kliniske overvejelser
Sygdomsassocieret moderlig og/eller føtal/neonatal risiko
PNH under graviditet er forbundet med ugunstige moderlige resultater, herunder forværring af cytopenier trombotiske begivenheder infektioner blødende aborter og øget mødredødelighed og ugunstige føtal resultater, herunder føtal død og for tidlig fødsel.
I graviditet er AHUS forbundet med ugunstige moderlige resultater inklusive Preeclampsia og preterm delivery og adverse fetal/neonatal outcomes including intrauterine growth restriction (IUGR) fetal death og low birth weight.
Data
Dyredata
Dyreproduktionsundersøgelser blev udført i mus under anvendelse af doser af et murint anti-C5-antistof, der tilnærmede 1-2,2 gange (belastningsdosis) og 0,8-1,8 gange (vedligeholdelsesdosis) den anbefalede humane ultomiris-dosis baseret på en kropsvægt sammenligning. Når dyreeksponering for antistoffet forekom i tidsperioden fra før parring, indtil tidlig drægtighed ikke blev observeret noget fald i fertilitet eller reproduktiv ydeevne. Når moderens eksponering for antistoffet forekom under organogenese 2 tilfælde af nethindens dysplasi og 1 tilfælde af navlestrengsbrok blev observeret blandt 230 afkom født af mødre udsat for den højere antistofdosis; Eksponeringen øgede imidlertid ikke føtaltab eller neonatal død. Når moderens eksponering for antistoffet forekom i tidsperioden fra implantation gennem fravænning af et højere antal mandlige afkom blev moribund eller døde (1/25 kontroller 2/25 lav dosisgruppe 5/25 høj dosisgruppe). Overlevende afkom havde normal udvikling og reproduktiv funktion. Human IgG er kendt for at krydse den humane placentale barriere, og dermed kan ultomiris potentielt forårsage terminalkomplementinhibering i føtal cirkulation.
Amning
Risikooversigt
Der er ingen data om tilstedeværelsen af ravulizumab-cwvz i human mælk effekten på det ammede barn eller effekten på mælkeproduktionen. Da mange medicinske produkter og immunoglobuliner udskilles i human mælk, og på grund af potentialet for alvorlige bivirkninger i et ammende barn, bør amning afbrydes under behandlingen og i 8 måneder efter den endelige dosis.
Pædiatrisk brug
Sikkerheden og effektiviteten af Ultomiris til behandling af PNH er blevet etableret hos pædiatriske patienter i en måned og ældre. Brug af ultomiris til denne indikation understøttes af bevis fra tilstrækkelige og velstyrede forsøg hos voksne med yderligere farmakokinetisk effektivitet og sikkerhedsdata hos pædiatriske patienter i alderen 9 til 17 år [se Bivirkninger Klinisk farmakologi og Kliniske studier ]. The safety og efficacy feller the treatment of pediatric og adult patients with PNH appear similar. Use of Ultomiris in pediatric patients with PNH aged less than 9 years og body weight <30 kg is based on extrapolation of pharmacokinetic / pharmacodynamic (PK/PD) og efficacy og safety data from aHUS og PNH clinical studies [see Klinisk farmakologi og Kliniske studier ].
Sikkerheden og effektiviteten af Ultomiris til behandling af AHUS er blevet etableret hos pædiatriske patienter i en måned og ældre. Brug af ultomiris til denne indikation understøttes af bevis fra tilstrækkelige og velstyrede undersøgelser hos voksne med yderligere farmakokinetisk sikkerhed og effektivitetsdata hos pædiatriske patienter i alderen 10 måneder til <17 years. The safety og efficacy of Ultomiris feller the treatment of aHUS appear similar in pediatric og adult patients [see Bivirkninger og Kliniske studier ].
Sikkerheden og effektiviteten af Ultomiris til behandling af GMG eller NMOSD hos pædiatriske patienter er ikke blevet fastlagt.
Geriatrisk brug
Kliniske undersøgelser af ultomiris inkluderede ikke tilstrækkeligt antal forsøgspersoner i alderen 65 år og derover (58 patienter med PNH 9 med AHUS 54 med GMG og 7 med NMOSD) til at bestemme, om de reagerer anderledes end yngre forsøgspersoner.
DMG -supplement
Andre rapporterede kliniske erfaringer har ikke identificeret forskelle i responser mellem ældre og yngre patienter.
Overdoseringsoplysninger til Ultomiris
Ingen oplysninger leveret
Kontraindikationer for ultomiris
Ultomiris is contraindicated feller initiation in patients with unresolved serious Neisseria meningitidis infection [see ADVARSELS AND FORHOLDSREGLER ].
Klinisk farmakologi feller Ultomiris
Handlingsmekanisme
Ravulizumab-CWVZ er en terminal komplementinhibitor, der specifikt binder til komplementproteinet C5 med høj affinitet, hvilket hæmmer dets spaltning til C5A (den proinflammatoriske anafylatoxin) og C5B (den initierende underenhed af membranangrebskomplekset [MAC eller C5B-9]) således forhindrer MAC-formation. Ultomiris inhiberer terminal komplement-medieret intravaskulær hæmolyse hos patienter med PNH og komplement-medieret thrombotisk mikroangiopati (TMA) hos patienter med AHUS.
Den nøjagtige mekanisme, hvormed Ravulizumab-CWVZ udøver sin terapeutiske virkning hos GMG-patienter, er ukendt, men antages at involvere reduktion af terminal komplementkompleks C5B-9-afsætning ved det neuromuskulære kryds.
Den nøjagtige mekanisme, hvormed Ravulizumab-CWVZ udøver sin terapeutiske virkning i NMOSD, er ukendt, men antages at involvere inhibering af aquaporin-4 antistofinduceret terminalkomplement C5B-9-afsætning.
Farmakodynamik
Komplet inhibering af serumfri C5 (koncentration på mindre end 0,5 mcg/ml) blev observeret ved afslutningen af den første Ultomiris-infusion og opretholdt gennem hele 26-ugers behandlingsperiode hos både voksne og pædiatriske patienter med PNH hos størstedelen (93%) af voksne og pædiatriske patienter med AHUS hos alle voksne patienter med GMG og i størstedelen (98,3%) af voksne patienter med NMOSD.
Omfanget og varigheden af den farmakodynamiske respons hos patienter med PNH AHUS GMG eller NMOSD var eksponeringsafhængig for Ultomiris. Gratis C5 -niveauer af <0.5 mcg/mL were cellerrelated with maximal intravascular hemolysis control og complete terminal complement inhibition in patients with PNH.
Komplet terminalkomplementinhibering efter initiering af ultomiris-behandling førte til normalisering af serum LDH i uge 4 i komplementinhibitor naive patienter med PNH og vedligeholdt LDH-normalisering hos patienter, der tidligere blev behandlet med eculizumab med PNH [se Kliniske studier ].
Farmakokinetik
Følgende Ultomiris-behandling øges ravulizumab-CWVZ-farmakokinetik forholdsmæssigt over et dosisområde fra 200 til 5400 mg. Ravulizumab-CWVZ Cmax og Ctrough-parametre er vist i tabel 17 Tabel 18 Tabel 19 og tabel 20.
Tabel 17: Gennemsnit (%CV) Farmakokinetiske parametre efter Ultomiris-behandling hos patienter med PNH, der er komplementinhibitor-naive og patienter, der tidligere blev behandlet med eculizumab
| Pædiatriske patienter | Voksne patienter | ||||||||
| ALXN1210-PNH-304 | ALXN1210-PNH- 301 | ALXN1210-PNH- 302 | |||||||
| N | Komplementinhibitor- naiv | N | Tidligere behandlet med eculizumab | N | Komplementinhibitor- naiv | N | Tidligere behandlet med eculizumab | ||
| Cmax (MCG/ML) | Ld | 4 | 733 (14.5) | 8 | 885 (19.3) | 125 | 771 (21.5) | 95 | 843 (24.1) |
| MD | 4 | 1490 (26.7) | 8 | 1705 (9.7) | 124 | 1379 (20.0) | 95 | 1386 (19.4) | |
| Ctrough (MCG/ML) | Ld | 4 | 368 (14.7) | 8 | 452 (15.1) | 125 | 391 (35.0) | 96 | 405 (29.9) |
| MD | 4 | 495 (21.3) | 8 | 566 (12.2) | 124 | 473 (33.4) | 95 | 501 (28.6) | |
| Forkortelser: LD = indlæsning af dosis; MD = vedligeholdelsesdosis |
Tabel 18: Gennemsnit (%CV) Farmakokinetiske parametre efter Ultomiris -behandling hos patienter med AHUS
| Pædiatriske patienter (ALXN1210-AHUS-312) | Voksne patienter (ALXN1210-AHUS-311) | ||||||
| N | <20 kg MD Q4W | N | ≥ 20 til <40 kg MD Q8W | N | ≥ 40 kg MD Q8W | ||
| Cmax (MCG/ML) | Ld | 8 | 656 (38.1) | 4 | 600 (17.3) | 52 | 754 (35.2) |
| MD | 7 | 1467 (37.8) | 6 | 1863 (15.3) | 46 | 1458 (17.6) | |
| Ctrough (MCG/ML) | Ld | 9 | 241 (52.1) | 5 | 186 (16.5) | 55 | 313 (33.9) |
| MD | 7 | 683 (46.1) | 6 | 549 (34.1) | 46 | 507 (42,5) | |
| Forkortelser: LD = indlæsning af dosis; MD = vedligeholdelsesdosis; Q4W = Every 4 Weeks; Q8W = Every 8 Weeks |
Tabel 19: Gennemsnit (%CV) Farmakokinetiske parametre efter Ultomiris -behandling hos voksne patienter med GMG
| N | Voksne patienter (ALXN1210-MG-306) | ||
| Cmax (MCG/ML) | Ld | 86 | 874 (21.1) |
| MD | 76 | 1548 (23.2) | |
| Ctrough (MCG/ML) | Ld | 85 | 418 (27.6) |
| MD | 70 | 587 (29.6) | |
| Forkortelser: LD = indlæsning af dosis; MD = vedligeholdelsesdosis |
Tabel 20: Gennemsnit (%CV) Farmakokinetiske parametre efter Ultomiris -behandling hos voksne patienter med NMOSD
| N | Voksne patienter (ALXN1210-NMO-307) | ||
| Cmax (MCG/ML) | Ld | 58 | 935.3 (17.3) |
| MD | 56 | 1836.4 (19.4) | |
| Ctrough (MCG/ML) | Ld | 58 | 459.1 (19.7) |
| MD | 54 | 796.9 (27.1) | |
| Forkortelser: LD = indlæsning af dosis; MD = vedligeholdelsesdosis |
Fordeling
Den gennemsnitlige (standardafvigelse [SD]) distributionsvolumen ved stabil tilstand hos patienter med PNH AHUS GMG eller NMOSD er vist i tabel 21.
Eliminering
Den gennemsnitlige (standardafvigelse [SD]) terminal eliminering halveringstid og clearance af Ravulizumab-CWVZ er vist i tabel 21.
Tabel 21: Distribution Biotransformation og elimineringsparametre efter Ultomiris -behandling
| Voksne og pædiatriske patienter med PNH | Voksne og pædiatriske patienter med AHUS | Voksne patienter with gMG | Voksne patienter with NMOSD | |
| Fordeling | ||||
| Distributionsvolumen i stabil tilstand (liter) gennemsnit (SD) | 5.30 (0.95) | 5.22 (1.85) | 5.74 (1.16) | 4.77 (0.819) |
| Biotransformation og eliminering | ||||
| Terminal eliminering halveringstid (dage) middelværdi (SD) | 49.6 (9.08) | 51.8 (16.2) | 56.6 (8.36) | 64.3 (11.0) |
| Clearance (liter/dag) middelværdi (SD) | 0,08 (0,02) | 0,08 (0,04) | 0,08 (0,02) | 0,05 (0,016) |
Specifikke populationer
Der blev ikke observeret klinisk signifikante forskelle i farmakokinetikken i Ravulizumab-CWVZ baseret på kønsalder (10 måneder til 83 år) racehepatisk svækkelse eller nogen grad af nyrefunktion inklusive patienter med proteinuri eller modtagelse af dialyse.
Kropsvægt var et klinisk signifikant covariat på farmakokinetikken af Ravulizumab-CWVZ.
Lægemiddelinteraktioner
Der er ikke udført nogen interaktionsundersøgelser med lægemiddel.
Neonatal FC -receptorblokkeringsbehandling kan forstyrre de endosomale neonatale FCRN -genvindingsmekanisme for monoklonale antistoffer, såsom ravulizumab og derved reducere serum ravulizumab -koncentrationer [se Lægemiddelinteraktioner ].
Samtidig PE PP eller IVIG -behandling kræver en supplerende dosis af ultomiris [se Dosering og administration ].
Immunogenicitet
Den observerede forekomst af anti-lægemiddelantistoffer er meget afhængig af følsomheden og specificiteten af assayet. Forskelle i analysemetoder udelukker meningsfulde sammenligninger af forekomsten af anti-drug-antistoffer i undersøgelserne beskrevet nedenfor med forekomsten af anti-lægemiddelantistoffer i andre undersøgelser, herunder dem af Ultomiris eller andre Ravulizumab-CWVZ-produkter.
Immunogeniciteten af Ravulizumab-CWVZ er blevet evalueret under anvendelse af et enzymbundet immunosorbentassay (ELISA) til påvisning af binding af anti-ravulizumab-CWVZ-antistoffer. For patienter, hvis sera testede positivt i screeningsimmunoassayet, blev der udført in vitro -biologisk assay for at påvise neutraliserende antistoffer.
I kliniske studier med Ultomiris-behandlingsvækst-antistoffer mod Ravulizumab-CWVZ blev detekteret hos 1 ud af 219 (0,5%) patienter med PNH [se Kliniske studier ] og 1 ud af 71 (1,4%) patienter med AHUS [se Kliniske studier ]. In these 2 patient populations the observed ADA were non-neutralizing with no apparent impact on PK safety eller efficacy. In the gMG study (N = 86) og NMOSD study (N = 58) no treatment-emergent antibodies to ravulizumab-cwvz were detected [see Kliniske studier ].
Imidlertid er assayet, der blev anvendt til at måle anti-lægemiddelantistoffer (ADA), underlagt interferens af serum ravulizumab-CWVZ, der muligvis resulterer i en undervurdering af forekomsten af antistofdannelse. På grund af begrænsningen af assaybetingelserne vides den potentielle kliniske virkning af antistoffer for Ravulizumab-CWVZ ikke.
Kliniske studier
Paroxysmal natlig hæmoglobinuri (PNH)
Sikkerheden og effektiviteten af Ultomiris hos voksne patienter med PNH blev vurderet i 2 åben mærket randomiseret aktivkontrolleret ikke-inferioritetsfase 3-undersøgelser: PNH-undersøgelse 301 og PNH-undersøgelse 302. Undersøgelse 301 indskrevne patienter med PNH, der var komplementinhibitor naiv og havde aktiv hæmolyse. Undersøgelse 302 tilmeldte patienter med PNH, som var klinisk stabile efter at have været behandlet med eculizumab i mindst de sidste 6 måneder. Sikkerheden og effektiviteten af Ultomiris hos pædiatriske patienter med PNH blev vurderet i PNH-undersøgelse 304 En open-label fase 3-undersøgelse udført i eculizumab-erfarne og komplementinhibitorbehandling naive pædiatriske patienter med PNH.
I undersøgelse 301 og undersøgelse 302 voksne med PNH blev doseret med Ultomiris administreret intravenøst i overensstemmelse med den vægtbaserede dosering beskrevet i afsnit 2.3 (4 infusioner af Ultomiris over 26 uger) ovenfor. Eculizumab blev administreret på dage 1 8 15 og 22 efterfulgt af vedligeholdelsesbehandling med 900 mg eculizumab på dag 29 og hver 2. uge (Q2W) derefter for i alt 26 ugers behandling i henhold til den godkendte doseringsregime af eculizumab, som var standard-of-CARE for PNH på det tidspunkt i undersøgelserne.
Patienter blev vaccineret mod meningokokkinfektion før eller på tidspunktet for påbegyndelse af behandling med ultomiris eller eculizumab eller modtog profylaktisk behandling med passende antibiotika indtil 2 uger efter vaccination. Profylaktisk behandling med passende antibiotika ud over 2 uger efter vaccination var efter udbyderens skøn.
Undersøg i komplementinhibitor naive voksne patienter med PNH
Den komplement-inhibitor naive undersøgelse [ALXN1210-PNH-301; NCT02946463] var en 26-ugers multicenter open-label randomiseret aktivkontrolleret ikke-mindrevinionsfase 3-undersøgelse udført i 246 patienter naive for at komplementere inhibitorbehandling inden studieindgangen.
Patienter med PNH med flowcytometrisk bekræftelse af mindst 5% PNH -celler blev randomiseret 1: 1 til enten intravenøst administreret ultomiris eller eculizumab. Den gennemsnitlige samlede PNH -granulocytklonstørrelse var 85% den gennemsnitlige totale PNH -monocytklonstørrelse var 88%, og den gennemsnitlige samlede PNH RBC -klonstørrelse var 39%. Otteoghalvfjerds procent af patienterne havde en dokumenteret PNH-associeret tilstand, der blev diagnosticeret før tilmelding til forsøget: anæmi (85%) hæmoglobinuri (63%) historie med aplastisk anæmi (32%) historie med nyresvigt (12%) myelodysplastisk syndrom (5%) graviditetskomplikationer (3%) og andre (16%). Større baselineegenskaber blev afbalanceret mellem behandlingsgrupper. Tabel 22 tilvejebringer baselineegenskaberne for de patienter, der er indskrevet i den naive undersøgelse af komplementinhibitor.
Tabel 22: Baselineegenskaber i den naive undersøgelse af komplementinhibitor
| Parameter | Statistik | Ultomiris (N = 125) | Eculizumab (N = 121) |
| Alder (år) ved første infusion i studiet | Gennemsnit (SD) | 44.8 (15.2) | 46.2 (16.2) |
| Min maks | 18 83 | 18 86 | |
| Køn | |||
| Han | n (%) | 65 (52.0) | 69 (57.0) |
| Race | |||
| asiatisk | n (%) | 72 (57.6) | 57 (47.1) |
| Hvid | 43 (34.4) | 51 (42.1) | |
| Sort eller afroamerikaner | 2 (1.6) | 4 (3.3) | |
| Amerikansk indisk eller alaska indfødt | 1 (0,8) | 1 (0,8) | |
| Andre | 4 (3.2) | 4 (3.3) | |
| Ikke rapporteret | 3 (2.4) | 4 (3.3) | |
| Forbehandling LDH-niveauer (U/L) | Median | 1513.5 | 1445.0 |
| Min maks | (378.0 3759.5) | (423.5 3139.5) | |
| Enheder af PRBC/fuldblod | Median | 6.0 | 6.0 |
| overført inden for 12 måneder før | |||
| Første dosis | Min maks | (1 44) | (1 32) |
| Antithrombotiske midler, der bruges inden for 28 dage før den første dosis | n (%) | 22 (17.6) | 22 (18.2) |
| Patienter med en Mave History | n (%) | 17 (13.6) | 25 (20.7) |
| Patienter med en historie med trombose | n (%) | 17 (13.6) | 20 (16.5) |
| Patienter med samtidig antikoagulantbehandling | n (%) | 23 (18.4) | 28 (23.1) |
| Forkortelser: LDH = lactatdehydrogenase; max = maksimum; min = minimum; Mave = større ugunstig vaskulær begivenhed; PRBC = pakket røde blodlegemer; SD = standardafvigelse |
Effektivitet blev etableret baseret på overtrædelse af transfusion og hæmolyse som direkte målt ved normalisering af LDH -niveauer. Undgåelse af transfusion blev defineret som patienter, der ikke modtog en transfusion og ikke opfyldte de protokolspecificerede retningslinjer for transfusion fra baseline op til dag 183. Støttende effektivitetsdata omfattede den procentvise ændring fra basislinjen i LDH -niveauer. Andelen af patienter med gennembrudt hæmolyse defineret som mindst et nyt eller forværring af symptom eller underskrivning af intravaskulær hæmolyse i tilstedeværelsen af ellateret LDH ≥ 2 x x x x x x x x x x x x x x x x x x x x ≥ 2 x <1.5 x ULN on therapy og the propellertion of patients with stabilized hæmoglobin.
Ikke-mindreværd af Ultomiris over for eculizumab blev demonstreret på tværs af slutpunkter i den komplement-inhibitor naive behandlingspopulation beskrevet i tabel 23 nedenfor.
Tabel 23: Effektivitet resulterer i den naive undersøgelse af komplementinhibitor
| Ultomiris (N = 125) | Eculizumab (N = 121) | Statistik til sammenligning | Behandlingseffekt (95% CI) | |
| Overtrædelse af transfusion | 73,6% | 66,1% | Forskel i sats | 6.8 (-4.66 18.14) |
| LdH nellermalization | 53,6% | 49,4% | Oddsforhold | 1.19 |
| LdH percent change | -76,84% | -76,02% | Forskel i % ændring fra baseline | -0.83 (-5.21 3.56) |
| Gennembrud hæmolyse | 4,0% | 10,7% | Forskel i sats | -6.7 (-14,21 0,18) |
| Hæmoglobinstabilisering | 68,0% | 64,5% | Forskel i sats | 2.9 (-8,80 14,64) |
| For transfusionsundgåelse af endpoint -behandlingsforskelle (95% CI'er) er baseret på estimerede forskelle i procent med 95% CI. For laktat dehydrogenase -normaliseringsendet vises den justerede forekomst inden for hver behandling. Forkortelser: LDH = lactatdehydrogenase; CI = konfidensinterval |
Der var ingen observerbar forskel i træthed mellem ultomiris og eculizumab efter 26 ugers behandling sammenlignet med baseline målt ved facit-fatigue-instrumentet. Patientrapporteret træthed kan være en under- eller overdreven estimering, fordi patienter ikke var blændet for behandlingsopgaven.
Undersøgelse i eculizumab-erfarne voksne patienter med PNH
The study in eculizumab-experienced patients [ALXN1210-PNH-302; NCT03056040] var en 26-ugers multicenter open-label randomiseret aktivkontrolleret ikke-mindrevinær fase 3-undersøgelse udført i 195 patienter med PNH, som var klinisk stabile efter at have været behandlet med eculizumab i mindst de sidste 6 måneder.
Patienter, der demonstrerede klinisk stabil sygdom efter at have været behandlet med eculizumab i mindst de foregående 6 måneder, blev randomiseret 1: 1 for enten at fortsætte eculizumab eller for at skifte til ultomiris administreret intravenøst. Den gennemsnitlige samlede PNH -granulocytklonstørrelse var 83% den gennemsnitlige totale PNH -monocytklonstørrelse var 86%, og den gennemsnitlige samlede PNH RBC -klonstørrelse var 60%. Femoghalvfem procent af patienterne havde en dokumenteret PNH-associeret tilstand, der blev diagnosticeret inden tilmelding til forsøget: anæmi (67%) hæmaturi eller hæmoglobinuri (49%) historie med aplastisk anæmi (37%) historie med nyresvigt (9%) myelodysplastisk syndrom (5%) graviditetskomplikation (7%) og andre (14%). Større baselineegenskaber blev afbalanceret mellem de 2 behandlingsgrupper. Tabel 24 tilvejebringer baselineegenskaberne for de patienter, der er indskrevet i den eculizumab-erfarne undersøgelse.
Tabel 24: Baselineegenskaber i eculizumab-erfarne voksne patienter med PNH
| Parameter | Statistik | Ultomiris (N = 97) | Eculizumab (N = 98) |
| Alder (år) ved første infusion i studiet | Gennemsnit (SD) | 46.6 (14.41) | 48.8 (13.97) |
| Min maks | 18 79 | 23 77 | |
| Race | n (%) | 50 (51,5) | 61 (62.2) |
| Hvid | 23 (23.7) | 19 (19.4) | |
| asiatisk | 5 (5.2) | 3 (3.1) | |
| Sort eller afroamerikaner | 2 (2.1) | 1 (1.0) | |
| Andre | 13 (13.4) | 13 (13.3) | |
| Ikke rapporteret | 3 (3.1) | 1 (1.0) | |
| Ukendt multipel | 1 (1.0) | 0 | |
| Køn | |||
| Han | n (%) | 50 (51,5) | 48 (49.0) |
| Forbehandling LDH-niveauer (U/L) | Median Min maks | 224.0 135.0 383.5 | 234,0 100,0 365,5 |
| Enheder af PRBC/fuldblod overført inden for 12 måneder før Første dosis | Median | 4.0 | 2.5 |
| Min maks | (1 32) | (2 15) | |
| Antithrombotiske midler, der bruges inden for 28 dage før den første dosis | n (%) | 20 (20.6) | 13 (13.3) |
| Patienter med en Mave History | n (%) | 28 (28.9) | 22 (22.4) |
| Patienter med en historie med trombose | n (%) | 27 (27.8) | 21 (21.4) |
| Patienter med samtidig antikoagulantbehandling | n (%) | 22 (22.7) | 16 (16.3) |
| Forkortelser: LDH = lactatdehydrogenase; max = maksimum; min = minimum; Mave = større ugunstig vaskulær begivenhed; PRBC = pakket røde blodlegemer; SD = standardafvigelse |
Effektivitet blev fastlagt baseret på hæmolyse målt ved LDH -procent ændring fra baseline til dag 183, og understøttende effektivitetsdata var overtrædelse af overtrædelsesforsyning af patienter med stabiliseret hæmoglobin og andelen af patienter med gennembrudte hæmolyse gennem dag 183.
Ikke-mindreværd af ultomiris over for eculizumab blev påvist på tværs af slutpunkter hos patienterne med PNH, der tidligere var behandlet med eculizumab beskrevet i tabel 25 nedenfor.
Tabel 25: Effektivitet resulterer i eculizumab-erfarne voksne patienter med PNH eculizumab-erfarne undersøgelse
| Ultomiris N = 97 | Eculizumab N = 98 | Statistik til sammenligning | Behandlingseffekt (95% CI) | |
| LdH percent change | -0,82% | 8,4% | Forskel i % ændring fra baseline | 9.2 (-0.42 18.8) |
| Gennembrud hæmolyse | 0% | 5,1% | Forskel i sats | 5.1 (-8.9 19.0) |
| Undgåelse af transfusion | 87,6 % | 82,7% | Forskel i sats | 5.5 (-4.3 15.7) |
| Hæmoglobinstabilisering | 76,3% | 75,5% | Forskel i sats | 1.4 (-10.4 13.3) |
| Forkortelser: CI = konfidensinterval; LDH = lactatdehydrogenase |
Der var ingen observerbar forskel i træthed mellem ultomiris og eculizumab efter 26 ugers behandling sammenlignet med baseline målt ved facit-fatigue-instrumentet. Patientrapporteret træthed kan være en under-eller-overdreven estimering, fordi patienter ikke var blændet for behandlingsopgaven.
Undersøgelse hos eculizumab-erfarne og komplementinhibitor naive pædiatriske patienter med PNH
Den pædiatriske undersøgelse ALXN1210-PNH-304 (NCT03406507) var en multicenter åben mærket fase 3-undersøgelse udført i eculizumab-eksperienced og komplementinhibitorbehandlingsnaive pædiatriske patienter med PNH. I alt 13 pædiatriske patienter med PNH afsluttede intravenøst administreret ultomiris -behandling i den primære evalueringsperiode (26 uger). Fem af de 13 patienter var aldrig blevet behandlet med komplementinhibitorer, og 8 patienter blev behandlet med eculizumab. Elleve af de tretten patienter var mellem 12 og 17 år ved første infusion med 2 patienter under 12 år (11 og 9 år gamle). Tabel 26 viser basislinjekarakteristika for de pædiatriske patienter, der er indskrevet i undersøgelse Alxn1210-PNH-304.
Tabel 26: Baselineegenskaber for pædiatriske patienter med PNH
| Variabel | Komplementinhibitorbehandlingsnaive patienter (N = 5) | Eculizumab- Experienced Patients (N = 8) | Alle patienter (N = 13) |
| Køn n (%) | |||
| Han | 4 (80.0) | 1 (12.5) | 5 (38.5) |
| Kvinde | 1 (20.0) | 7 (87,5) | 8 (61,5) |
| Alder ved første infusion (år) | |||
| Gennemsnit (SD) | 14.4 (2.2) | 14.4 (3.1) | 14.4 (2.7) |
| Median (min max) | 15.0 (11 17) | 15.0 (9 17) | 15.0 (9 17) |
| Alder ved første infusion (år) categellery n (%) | |||
| <12 years | 1 (20.0) | 1 (12.5) | 2 (15.4) |
| ≥ 12 år | 4 (80.0) | 7 (87,5) | 11 (84.6) |
| Baselinevægt (kg) | |||
| Gennemsnit (SD) | 56.3 (11.6) | 56.3 (12.2) | 56.3 (11.5) |
| Median (min max) | 55.6 (39.5 72.0) | 55,5 (36,7 69,0) | 55,6 (36,7 72,0) |
| Baselinevægt (kg) categellery n (%) | |||
| ≥ 30 til <40 kg | 1 (20.0) | 1 (12.5) | 2 (15.4) |
| ≥ 40 til <60 kg | 3 (60.0) | 4 (50.0) | 7 (53.8) |
| ≥ 60 til <100 kg | 1 (20.0) | 3 (37,5) | 4 (30.8) |
| Enheder af PRBC/fuldblod overført inden for 12 måneder før Første dosis | |||
| Median (min max) | 7.0 (3 11) | 2.0 (2 2) | - |
| Forbehandling LDH-niveauer (U/L) | |||
| Median (min max) | 588.5 (444 2269.7) | 251,5 (140,5 487) | - |
| Bemærk: Procentdelene var baseret på det samlede antal patienter i hver kohort eller samlet. Forkortelser: LDH = lactatdehydrogenase; kg = kg; max = maksimum; min = minimum; PRBC = pakket røde blodlegemer; SD = standardafvigelse |
Baseret på kropsvægt, modtog patienter en belastningsdosis af ultomiris på dag 1 efterfulgt af vedligeholdelsesbehandling på dag 15 og en gang hver 8. uge (Q8W) derefter for patienter, der vejer ≥ 20 kg eller en gang hver 4. uge (Q4W) for patienter, der vejer <20 kg. Feller patients who entered the study on eculizumab therapy Day 1 of study treatment was planned to occur 2 weeks from the patient's last dose of eculizumab.
Den vægtbaserede dosisregime af Ravulizumab-CWVZ tilvejebragte inhibering af terminalkomplement hos alle patienter i hele 26-ugers behandlingsperiode uanset forudgående erfaring med eculizumab. Efter påbegyndelse af Ravulizumab-CWVZ-behandling blev den terapeutiske serumkoncentrationer af Ravulizumab-CWVZ opnået efter den første dosis og opretholdt i hele den primære evalueringsperiode i begge kohorter. Tre af 5 komplementinhibitorbehandlingsnaive patienter og 6 ud af 8 eculizumab-erfarne patienter opnåede henholdsvis hæmoglobinstabilisering i henhold til uge 26. Undgåelse af transfusion blev nået til 11 ud af 13 af patienterne i den 26-ugers primære evalueringsperiode. En patient oplevede gennembrud hæmolyse i forlængelsesperioden. Tabel 27 viser sekundære effektivitetsresultater for den primære evalueringsperiode.
Tabel 27: Effektivitetsresultater fra den 26-ugers primære evalueringsperiode for pædiatrisk patientundersøgelse i PNH (ALXN1210-PNH-304)
| Slutpunkt | Behandling naiv (N = 5) | Eculizumab Experienced (N = 8) |
| LdH percent change from baseline (%)a | -47.9 (-113.4 17.5) | 4.7 (-36,7 46,0) |
| Undgåelse af transfusion (%)b | 60,0 (14,7 94,7) | 100,0 (63,1 100,0) |
| Skift make-fatiguea | 3.4 (-4.2 11.0) | 1.3 (-3.1 5.7) |
| Hæmoglobinstabilisering (%)b | 60,0 (14,7 94,7) | 75,0 (34,9 96,8) |
| Gennembrud hæmolyse (%) | 0 | 0c |
| a 95% CI'er for det gennemsnitlige opnåede fra T-fordeling blev præsenteret. b 95% CI'er for andelen var baseret på nøjagtige konfidensgrænser ved hjælp af Clopper-Pearson-metoden. c Ingen patienter oplevede gennembrud hæmolyse i den primære evalueringsperiode. En patient oplevede gennembrud hæmolyse efter 1,8 år i forlængelsesperioden; Men på tidspunktet for den gennembrudte hæmolysebegivenhed havde patienten tilstrækkelig C5 -hæmning (gratis C5 <0.5 mcg/mL). Abbreviations: FACIT = Functional Assessment of Chronic Illness Therapy; LdH = lactate dehydrogenase |
En klinisk relevant forbedring fra baseline i træthed som vurderet ved pædiatrisk facit-fatigue (dvs. gennemsnitlig forbedring af> 3 enheder til pædiatriske facit-træthedsresultater) blev opretholdt i hele den primære evalueringsperiode i 5-komplementinhibitorbehandling naive patienter. En lille forbedring blev også observeret hos eculizumab-eksperiderede patienter. Imidlertid kan patientrapporteret træthed være en under-eller-overdreven estimering, fordi patienter ikke var blændet for behandlingsopgaven.
Effektiviteten af Ultomiris hos pædiatriske patienter med PNH svarer til den, der blev observeret hos voksne patienter med PNH, der er indskrevet i pivotale studier.
Atypisk hæmolytisk uremisk syndrom (aHUS)
Effektiviteten af Ultomiris administreret intravenøst hos patienter med AHUS blev vurderet i 2 open-label enkeltarmsundersøgelser. Undersøg ALXN1210-AHUS-311 tilmeldte voksne patienter, der viste tegn på TMA. For at kvalificere sig til tilmelding blev patienter forpligtet til at have et blodpladetælling ≤ 150 x9/L Bevis for hæmolyse, såsom en forhøjelse i serum LDH og serumkreatinin over de øvre grænser for normal eller krævet dialyse.
Undersøg ALXN1210-AHUS-312 tilmeldte pædiatriske patienter, der viste tegn på TMA. For at kvalificere sig til tilmelding blev patienter forpligtet til at have et blodpladetælling ≤ 150 x9/L Bevis for hæmolyse, såsom en forhøjelse i serum LDH og serumkreatininniveau ≥ 97,5% percentil ved screening eller krævet dialyse. I begge undersøgelser til tilmeldingskriterier udelukkede patienter, der præsenterede med TMA på grund af en desintegrin og metalloproteinase med et thrombospondin-type 1-motivmedlem 13 (ADAMTS13) -mangel Shiga Toxin Escherichia coli-relateret hæmolytisk uremisk syndrom (stec-Hus) og genetisk defekt i Cobalamin C metabolisme. Patienter med bekræftet diagnose af stec-Hus efter tilmelding blev udelukket fra effektivitetsevalueringen.
Undersøg hos voksne patienter med AHUS
Den voksne undersøgelse [ALXN1210-AHUS-311; NCT02949128] blev udført hos patienter, der var naive for at supplere inhibitorbehandling før undersøgelsesindgangen. Undersøgelsen bestod af en 26-ugers første evalueringsperiode, og patienter fik lov til at gå ind i en forlængelsesperiode i op til 4,5 år. Alle patienter modtog ultomiris administreret intravenøst i henhold til deres vægt [se Dosering og administration ].
I alt 56 patienter med AHUS blev evalueret for effektivitet. Treoghalvfjerds procent af patienterne havde ekstra-renal tegn (kardiovaskulær pulmonal centralnervesystem gastrointestinal hudskeletmuskel) eller symptomer på AHUS ved baseline. Ved baseline havde 71,4% (n = 40) af patienterne trin 5 kronisk nyresygdom (CKD). Fjorten procent havde en medicinsk historie med nyretransplantation, og 51,8% var på dialyse ved studieindgangen. Otte patienter gik ind i undersøgelsen med bevis for TMA i> 3 dage efter fødsel (dvs. postpartum).
Tabel 28 viser demografien og baselineegenskaberne for de 56 voksne patienter, der er indskrevet i studiet ALXN1210-AHUS-311, der udgjorde det fulde analysesæt.
Tabel 28: Demografi og baselineegenskaber i undersøgelsen AlXN1210-AHUS-311
| Parameter | Statistik | Ultomiris (N = 56) |
| Alder på tidspunktet for første infusion (år) | Gennemsnit (SD) | 42.2 (14.98) |
| Min maks | 19,5 76,6 | |
| Køn | ||
| Kvinde | n (%) | 37 (66.1) |
| Race a | n (%) | |
| Hvid | 29 (51.8) | |
| asiatisk | 15 (26.8) | |
| Ukendt | 8 (14.3) | |
| Andre | 4 (7.1) | |
| Blodplader (109/L) blod | n | 56 |
| [Normal interval 130 til 400 x 109/L] | Median (minmax) | 95.25 (18 473) |
| Hemoglobin (G/L) blod | n | 56 |
| [Normal interval 115 til 160 g/l (hun) 130 til 175 g/l (mand)] | Median (minmax) | 85,00 (60,5 140) |
| LdH (U/L) serum [nellermal range 120 to 246 U/L] | n | 56 |
| Median (minmax) | 508,00 (229,5 3249) | |
| EGFR (ml/min/1,73 m²) [Normalt interval ≥ 60 ml/min/1,73 m²] | n (%) | 55 |
| Gennemsnit (SD) | 15.86 (14.815) | |
| Median (minmax) | 10.00 (4 80) | |
| Bemærk: Procentdel er baseret på det samlede antal patienter. a Patienter kan have flere racer valgt. Forkortelser: EGFR = estimeret glomerulær filtreringshastighed; LDH = lactatdehydrogenase; max = maksimum; min = minimum; SD = standardafvigelse |
Effektivitetsevalueringen var baseret på komplet TMA-respons i den 26-ugers indledende evalueringsperiode, som det fremgår af normalisering af hæmatologiske parametre (blodpladeantal og LDH) og ≥ 25% forbedring i serumkreatinin fra baseline. Patienter måtte opfylde hvert komplette TMA -responskriterier ved 2 separate vurderinger opnået mindst 4 uger (28 dage) fra hinanden og enhver måling derimellem.
Komplet TMA-respons blev observeret hos 30 af de 56 patienter (54%) i den 26-ugers indledende evalueringsperiode som vist i tabel 29.
Tabel 29: Effektivitet resulterer i AHUS i den 26-ugers indledende evalueringsperiode (ALXN1210-AHUS-311)
| Total | Responder | ||
| n | Andel (95% CI)a | ||
| Komplet TMA -svar | 56 | 30 | 0.54 (0,40 0,67) |
| Komponenter i komplet TMA -respons blodpladetælling normalisering | 56 | 47 | 0.84 (0,72 0,92) |
| LdH nellermalization | 56 | 43 | 0.77 (0,64 0,87) |
| ≥ 25% forbedring i serumkreatinin fra baseline | 56 | 33 | 0.59 (0,45 0,72) |
| Hæmatologisk normalisering | 56 | 41 | 0.73 (0,60 0,84) |
| a 95% CI'er for andelen var baseret på nøjagtige konfidensgrænser ved hjælp af Clopper-Pearson-metoden. Forkortelser: CI = konfidensinterval; LDH = lactatdehydrogenase; TMA = thrombotic  microangiopathy |
En yderligere patient havde en komplet TMA-respons, der blev bekræftet efter den 26-ugers indledende evalueringsperiode. Komplet TMA -respons blev opnået på en median tid på 86 dage (rækkevidde: 7 til 169 dage). Den medianvarighed af komplet TMA -respons var 7,97 måneder (rækkevidde: 2,52 til 16,69 måneder). Alle svar blev opretholdt gennem al tilgængelig opfølgning.
bivirkninger af bystolisk 5 mg
Andre endpoints included platelet count change from baseline dialysis requirement og renal function as evaluated by estimated glomerular filtration rate (eGFR).
En stigning i gennemsnitlig blodpladetælling blev observeret efter påbegyndelse af ultomiris -behandlingen stigende fra 118.52 x9/L ved baseline til 240,34 x109/L på dag 8 og forbliver over 227 x9/L ved alle efterfølgende besøg i den indledende evalueringsperiode (26 uger).
Nyrefunktion målt ved EGFR blev forbedret eller opretholdt under Ultomiris -terapi. Den gennemsnitlige EGFR ( /-SD) steg fra 15,86 (14,82) ved baseline til 51,83 (39,16) med 26 uger. Hos patienter med komplet TMA -respons -nyrefunktion blev fortsat forbedret, efter at den komplette TMA -respons blev opnået.
Sytten af de 29 patienter (59%), der krævede dialyse ved undersøgelsesindgangen, ophørte dialyse ved udgangen af den tilgængelige opfølgning, og 6 af 27 (22%) patienter var slukket for dialyse ved baseline var på dialyse til sidst tilgængelig opfølgning.
Undersøg hos pædiatriske patienter med AHUS
Den pædiatriske undersøgelse [ALXN1210-AHUS-312; NCT03131219] er en 26-ugers igangværende multicenter-enkeltarmundersøgelse udført hos 16 pædiatriske patienter. Alle patienter modtog ultomiris administreret intravenøst i henhold til deres vægt [se Dosering og administration ]
I alt 14 eculizumab-naive patienter med dokumenteret diagnose af AHUS blev tilmeldt og inkluderet i denne foreløbige analyse. Medianalderen på tidspunktet for den første infusion var 5,2 år (område 0,9 17,3 år). Den samlede gennemsnitlige vægt ved baseline var 19,8 kg; Halvdelen af patienterne var i kategorien baselinevægt ≥ 10 til <20 kg. The majellerity of patients (71%) had pretreatment extra-renal signs (cardiovascular pulmonary central nervous system gastrointestinal skin skeletal muscle) eller symptoms of aHUS at baseline. At baseline 35.7% (n = 5) of patients had a CKD Stage 5. Seven percent had histellery of prieller kidney transplant og 35.7% were on dialysis at study entry.
Tabel 30 viser basislinjekarakteristika for de pædiatriske patienter, der er indskrevet i undersøgelsen ALXN1210-AHUS-312.
Tabel 30: Demografi og baselineegenskaber i undersøgelsen Alxn1210-ahus-312
| Parameter | Statistik | Ultomiris (N = 14) |
| Alder på tidspunktet for første infusion (år) categellery | n (%) | |
| Fødsel til <2 years | 2 (14.3) | |
| 2 til <6 years | 7 (50.0) | |
| 6 til <12 years | 4 (28.6) | |
| 12 til <18 years | 1 (7.1) | |
| Køn | n (%) | |
| Kvinde | 9 (64.3) | |
| Racea | n (%) | |
| Hvid | 7 (50.0) | |
| asiatisk | 4 (28.6) | |
| Sort eller afroamerikaner | 2 (14.3) | |
| Amerikansk indisk eller alaskan indfødt | 1 (7.1) | |
| Ukendt | 1 (7.1) | |
| Blodplader (109/L) blod [nellermal range 229 to 533 x 109/L] | Median (min max) | 64,00 (14 125) |
| Hemoglobin (G/L) blod [nellermal range 107 to 131 g/L] | ||
| Median (min max) | 74.25 (32 106) | |
| LdH (U/L) serum [nellermal range 165 to 395 U/L] | Median (min max) | 2077.00 (772 4985) |
| EGFR (ml/min/1,73 m²) [Normalt interval ≥ 60 ml/min/1,73 m²] | Gennemsnit (SD) | 28.4 (23.11) |
| Median (min max) | 22.0 (10 84) | |
| Bemærk: Procentdel er baseret på det samlede antal patienter. a Patienter kan have flere racer valgt. Forkortelser: EGFR = estimeret glomerulær filtreringshastighed; LDH = lactatdehydrogenase; max = maksimum; Min = minimum |
Effektivitetsevaluering var baseret på komplet TMA-respons i den 26-ugers indledende evalueringsperiode, som det fremgår af normalisering af hæmatologiske parametre (blodpladetælling og LDH) og ≥ 25% forbedring i serumkreatinin fra baseline. Patienter måtte opfylde alle komplette TMA -responskriterier ved 2 separate vurderinger opnået mindst 4 uger (28 dage) fra hinanden og enhver måling derimellem.
Komplet TMA -svar was observed in 10 of the 14 patients (71%) during the 26-week Initial Evaluation Period as shown in Table 31.
Tabel 31: Effektivitet resulterer i AHUS i den 26-ugers indledende evalueringsperiode (ALXN1210-AHUS-312)
| Total | Responder | ||
| n | Andel (95% CI)a | ||
| Komplet TMA -svar | 14 | 10 | 0,71 (0,42 0,92) |
| Komponenter i komplet TMA -respons blodpladetælling normalisering | 14 | 13 | 0,93 (0,66 0,99) |
| LdH nellermalization | 14 | 12 | 0,86 (0,57 0,98) |
| ≥ 25% forbedring i serumkreatinin fra baseline | 14 | 11 | 0,79 (0,49 0,95) |
| Hæmatologisk normalisering | 14 | 12 | 0,86 (0,57 0,98) |
| Bemærk: En patient trak sig tilbage fra undersøgelse efter modtagelse af 2 doser af Ravulizumab-CWVZ. a 95% CI'er for andelen var baseret på nøjagtige konfidensgrænser ved hjælp af Clopper-Pearson-metoden. Forkortelser: CI = konfidensinterval; LDH = lactatdehydrogenase; TMA = thrombotic microangiopathy |
Komplet TMA -svar during the Initial Evaluation Period was achieved at a median time of 30 days (range:15 to 88 days). The median duration of Komplet TMA -svar was 5.08 months (range: 3.08 to 5.54 months). All responses were maintained through all available follow-up.
Andre endpoints included platelet count change from baseline dialysis requirement og renal function as evaluated by eGFR.
En stigning i gennemsnitlig blodpladetælling blev observeret efter påbegyndelse af Ultomiris -behandlingen stigende fra 60,50 x 109/L ved baseline til 296,67 x9/L på dag 8 og forblev over 296 x9/L ved alle efterfølgende besøg i den indledende evalueringsperiode (26 uger). The mean eGFR (+/-SD) increased from 28.4 (23.11) at baseline to 108.0 (63.21) by 26 weeks.
Fire af de 5 patienter, der krævede dialyse ved undersøgelsesindgangen, var i stand til at afbryde dialyse efter den første måned i studiet og i varigheden af Ultomiris -behandling. Ingen patient startede dialyse under undersøgelsen.
Generaliserede Myasthenia Gravis (gMG)
Effektiviteten af Ultomiris til behandling af GMG blev påvist i en randomiseret dobbeltblind placebokontrolleret multicenterundersøgelse (ALXN1210-MG-306; NCT03920293). Patienter blev randomiseret 1: 1 for enten at modtage ultomiris (n = 86) eller placebo (n = 89) i 26 uger. Ultomiris blev administreret intravenøst i henhold til den vægtbaserede anbefalede dosering [se Dosering og administration ].
Patients with gMG with a positive serologic test for anti-AChR antibodies Myasthenia Gravis Foundation of America (MGFA) clinical classification class II to IV and Myasthenia Gravis-Activities of Daily Living (MG-ADL) total score ≥ 6 were enrolled. Baseline and disease characteristics were similar between treatment groups (including age at first dose [mean of 58 years for ULTOMIRIS versus 53 years for placebo] gender [51% female for ULTOMIRIS versus 51% female for placebo] race as White Asian and Black or African American [78% 17% and 2% for ULTOMIRIS versus 69% 18% and 5% for placebo respectively] and duration of MG since diagnosis [mean of 10 years ranging from 0.5 to 39.5 years for ULTOMIRIS versus mean of 10.0 years ranging from 0.5 to 36.1 years for placebo].
Over 80% af patienterne modtog acetylcholinesteraseinhibitorer 70% modtog kortikosteroider, og 68% modtog ikke-steroide immunsuppressiva (ISTS) ved studiepost. Patienter på samtidig medicin til behandling af GMG fik lov til at fortsætte med terapi i løbet af undersøgelsen.
Det primære effektendepunkt var en sammenligning af ændringen fra baseline mellem behandlingsgrupper i MG-ADL-samlede score i uge 26. MG-ADL er en kategorisk skala, der vurderer påvirkningen af den daglige funktion af 8 tegn eller symptomer, der typisk påvirkes i GMG. Hvert element vurderes på en 4-punkts skala, hvor en score på 0 repræsenterer normal funktion, og en score på 3 repræsenterer tab af evner til at udføre denne funktion. Den samlede score varierer fra 0 til 24 med de højere score, der indikerer mere svækkelse.
De sekundære endepunkter, der også blev vurderet fra basislinje til uge 26, omfattede ændringen i den kvantitative Mg Total Score (QMG). QMG er en kategorisk skala med 13 punkter, der vurderer muskelsvaghed. Hvert element vurderes på en 4-punkts skala, hvor en score på 0 ikke repræsenterer nogen svaghed, og en score på 3 repræsenterer alvorlig svaghed. En samlet score varierer fra 0 til 39, hvor højere score indikerer mere alvorlig svækkelse.
Andre secondary endpoints included the propellertion of patients with improvements of at least 5 og 3 points in the Qmg og Mg total scelleres respectively.
Behandling med ultomiris demonstrerede en statistisk signifikant ændring i MG-ADL og QMG total score fra baseline i uge 26 sammenlignet med placebo (tabel 32).
Tabel 32: Effektivitet resulterer hos patienter med GMG
| Effektivitet slutpunkter: Skift fra baseline i uge 26 | Placebo (n = 89) ls betyder | Ultomiris (n = 86) ls betyder | Behandlingseffekt (95% CI) | p-værdi* |
| Primært slutpunkt | ||||
| Mg | -1.4 | -3.1 | -1.6 (-2.6 -0.7) | <0.001 |
| Sekundært slutpunkt | ||||
| Qmg | -0.8 | -2.8 | -2.0 (-3.2 -0.8) | <0.001 |
| *P-værdi beregnet ved hjælp af blandet effektmodel til gentagne målinger Forkortelser: CI = konfidensinterval LS = mindst firkanter; Mg-adl = myasthenia gravis aktiviteter i dagligdagen; QMG = kvantitativ myasthenia gravis score for sygdomsgrad |
Andelen af QMG-respondenter med mindst en 5-punkts forbedring i uge 26 var større for ultomiris (30,0%) sammenlignet med placebo (11,3%) P = 0,005. Andelen af MG-ADL-respondenter med mindst en 3-punkts forbedring i uge 26 var også større for ultomiris (56,7%) sammenlignet med placebo (34,1%). Andelen af kliniske respondenter ved højere responstærskler (≥ 4- 5- 6- 7- eller 8-punkts forbedring af MG-ADL og ≥ 6- 7- 8- 9- eller 10-punkts forbedring af QMG) var konsekvent større for Ultomiris sammenlignet med placebo.
Figur 1: Skift fra baseline i MG-ADL Total score (A) og QMG Total score (B) til uge 26 i den randomiserede kontrollerede periode med ALXN1210-MG-306 (gennemsnit og 95% CI)
 |
Bemærk: *s<0.001 versus placebo
Neuromyelitis Optica Spectrum Disorder (NMOSD)
Effektiviteten og sikkerheden af Ultomiris hos voksne patienter med anti-AQP4-antistofpositive NMOSD blev vurderet i en open-label multicenter-undersøgelse AlxN1210-NMO-307 (NCT04291262). Patienter, der deltog i undersøgelse ALXN1210-NMO-307, modtog ultomiris intravenøst i den primære behandlingsperiode, der sluttede, da den sidst tilmeldte patient afsluttede (eller blev afbrudt før) 50 uger på undersøgelse, der repræsenterede en median studievarighed på 73,5 uger (mindst 13,7 maksimum 117,7). Effektivitetsvurderinger var baseret på en sammenligning af patienter i undersøgelse ALXN1210-NMO-307 med en ekstern placebo-kontrolgruppe fra en anden undersøgelse (undersøgelse ECU-NMO-301 NCT01892345) sammensat af en sammenlignelig population af voksne patienter med anti-AQP4-antistofpositive NMOSD.
Undersøg ALXN1210-NMO-307 tilmeldte 58 voksne patienter med NMOSD, der havde en positiv serologisk test for anti-AQP4-antistoffer mindst 1 tilbagefald i de sidste 12 måneder før screeningsperioden, og en udvidet handicapstatusskala (EDS) score ≤ 7. I de eksterne placebo-kontrolgruppe Kvalificeringskriterier var det lignende, bortset fra at patienterne var påkrævet at have mindst 2 tilnærmelser i de sidste 12 måneder. Sidste 24 måneder med mindst 1 tilbagefald i de 12 måneder før screeningen. Tidligere behandling med immunsuppressive terapier (ISTS) var ikke påkrævet til tilmelding. Imidlertid fik patienter på udvalgte IST'er (dvs. kortikosteroider azathioprin mycophenolat mofetil methotrexat og tacrolimus) tilladt at fortsætte med terapi med et krav til stabil dosering, indtil de nåede uge 106 i undersøgelsen. Lignende ist -brug var tilladt i den eksterne placebo -kontrolgruppe.
Ultomiris was administered intravenously accellerding to the weight-based recommended dosage [see Dosering og administration ].
Demografien var ens mellem Ultomiris-behandlingsgruppen fra undersøgelse ALXN1210-NMO-307 og placebo-behandlingsgruppen fra undersøgelse ECU-NMO-301 (inklusive alder [median på 46,0 år for Ultomiris versus 44,0 år for placebo] og køn [89,7% kvindelig for ultomiris versus 89,4% kvindelige for placering af placering Størstedelen af patienterne var hvide eller asiatiske. Mediantiden fra diagnose til den første dosis var 0,9 år for ultomiris og 2,0 år for placebo. Den median årlige tilbagefaldshastighed (ARR) i de sidste 24 måneder var 1,4 for Ultomiris versus 1,9 for placebo og det median antal historiske tilbagefald var 2 for Ultomiris versus 4 for placebo. Den median baseline EDSS -score var 3,3 for Ultomiris versus 4.0 for placebo. Ved baseline modtog 48% af patienterne i Ultomiris -gruppen samtidig ist inklusive kortikosteroider mod 72% af forsøgspersoner i placebogruppen.
Det primære slutpunkt for studiet ALXN1210-NMO-307 var tiden til først at dømme tilbagefald til retssagen som bestemt af et uafhængigt dommerudvalg. Der blev ikke observeret nogen retssag til retssag, hos Ultomiris-behandlede patienter i den primære behandlingsperiode, der repræsenterede en statistisk signifikant forskel mellem Ultomiris og placebo-behandlingsarme i tide til først at dømme retssagen (P <0.0001). The hazard ratio (95% confidence interval [CI]) feller Ultomiris compared with placebo was 0.014 (0.000 0.103) representing a 98.6% reduction in the risk of relapse (Figure 2). Ultomiris-treated patients experienced similar improvement in time to first adjudicated on-trial relapse with eller without concomitant treatment.
Figur 2: Kaplan-meier overlevelsesestimater for tid til først at dømme retssagen i undersøgelsen i studiet ALXN1210-NMO-307 og komparativ placebo-arm af studiet ECU-NMO-301
 |
Bemærk: Placebo-gruppedataene blev indsamlet som en del af undersøgelsen ECU-NMO-301. Patienter, der ikke oplevede en bedømmelse af retssagen, blev censureret i slutningen af undersøgelsesperioden. Hvis en patient i placebogruppen blev fulgt længere end nogen af patienterne i Ultomiris-gruppen, blev denne patient censureret ved den længste ultomiris opfølgningstid.
Patientinformation til Ultomiris
Ingen oplysninger leveret. Se ADVARSELS AND FORHOLDSREGLER afsnit.